
Syndrom úplné androgenní insenzitivity – raritní kazuistika malignizace dysgenetických gonád
Complete androgen insensitivity syndrome – rare case of malignancy of dysgenetic gonads
Objective: A case report of a young patient with primary amenorrhea who was diagnosed with agenesis of the uterus and was genetically confirmed for complete androgen insensitivity syndrome with already developed malignancy of dysgenetic gonads. Case report: The 17-year-old patient visited a gynecological clinic for primary amenorrhea. Both ultrasound and vaginal examination revealed suspicion of uterine agenesis, which was subsequently verified during diagnostic laparoscopy. Genetic testing showed karyotype 46,XY, and a rare diagnosis – complete androgen insensitivity syndrome. A secondary finding from a left gonadal biopsy was a Sertoli-Leydig cell tumor. The patient underwent bilateral gonadectomy and was given estrogen replacement therapy. She is now regularly examined by a pediatric oncologist. Conclusion: Complete androgen insensitivity syndrome is a rare genetic disease characterized by varying degrees of feminization in individuals with a male karyotype. It should not be neglected, especially in the differential diagnostic work-up of primary amenorrhea. Genetic testing of the karyotype should be performed whenever uterine agenesis is suspected.
Keywords:
androgen insensitivity syndrome – testicular feminization syndrome – androgen receptor – primary amenorrhea – Sertoli-Leydig cell tumor
Autoři:
V. Gamcová 1
![]() ; J. Eim 1
; J. Eim 1
![]() ; I. Meixnerová 2
; I. Meixnerová 2
![]() ; R. Hudeček 2
; R. Hudeček 2
![]()
Působiště autorů:
Gynekologicko-porodnické oddělení, Nemocnice Vyškov, p. o.
1; Gynekologicko-porodnická klinika LF MU a FN Brno
2
Vyšlo v časopise:
Ceska Gynekol 2022; 87(3): 184-187
Kategorie:
Kazuistika
doi:
https://doi.org/10.48095/cccg2022184
Souhrn
Cíl práce: Popis případu mladé pacientky s primární amenoreou, u níž byla diagnostikována ageneze dělohy a geneticky potvrzen syndrom úplné androgenní insenzitivity s již rozvinutou malignizací dysgenetických gonád. Kazuistika: Pacientka ve věku 17 let se dostavila do gynekologické ambulance pro primární amenoreu. Při ultrazvukovém i vaginálním vyšetření bylo vysloveno podezření na agenezi dělohy, které bylo následně verifikováno při diagnostické laparoskopii. Genetické vyšetření prokázalo karyotyp 46,XY a vzácnou diagnózu – syndrom úplné androgenní insenzitivity. Vedlejším nálezem z biopsie levé gonády byl tumor ze Sertoliho-Leydigových buněk. Pacientka podstoupila oboustrannou gonadektomii a byla jí nasazena substituční estrogenní terapie. Nyní je pravidelně dispenzarizována u dětského onkologa. Závěr: Syndrom úplné androgenní insenzitivity je vzácné genetické onemocnění projevující se různým stupněm feminizace u jedinců s mužským karyotypem. Nemělo by být opomíjeno zejména při diferenciálně diagnostické rozvaze primární amenorey. Genetické vyšetření karyotypu by mělo být provedeno vždy, když je vysloveno podezření na agenezi dělohy.
Klíčová slova:
syndrom androgenní insenzitivity – syndrom testikulární feminizace – androgenní receptor – primární amenorea – tumor ze Sertoliho-Leydigových buněk
Úvod
Syndrom androgenní insenzitivity (AIS – androgen insensitivity syndrome), dříve nazývaný syndrom testikulární feminizace, je nejčastějším z mužských pseudohermafroditizmů. Projevuje se různým stupněm feminizace zevního genitálu, poruchami rozvoje sekundárních pohlavních znaků v pubertě a infertilitou u jedinců s karyotypem 46,XY. Poprvé byl popsán Morrisem v roce 1953. Incidence se pohybuje mezi 2 a 5 : 100 000 [1]. Jedná se o hereditární onemocnění vázané na X chromozom způsobující poruchu androgenního receptoru (AR – androgen receptor). Gen pro AR je lokalizován na proximální části dlouhého raménka X chromozomu a bylo popsáno více než 1 000 mutací tohoto genu [2]. Právě proto je AIS řazen k jedněm z nejvariabilnějších genových poruch. Podle stupně závažnosti feminizace genitálu se AIS rozděluje do tří stupňů [3]:
• mírný AIS (MAIS – mild androgen insensitivity syndrom) – genitál je mužský, ale v důsledku azoospermie nebo těžké oligozoospermie jsou jedinci infertilní.Může být přítomna i gynekomastie;
• parciální AIS (PAIS – parcial androgen insenzitivity syndrom) – zevní genitál má variabilně vyjádřenou abnormální obojetní formu – od penisu s hypospadií po vulvu s klitoromegalií;
• úplný AIS (CAIS – complete androgen insenzitivity syndrom) – ženský zevní genitál – fenotyp jedince je ženský, ale s mužským genotypem.
V patogenezi onemocnění hraje roli, jak již bylo zmíněno, defektní receptor pro androgeny. Přítomné gonády jsou varlata, která mají přibližně normální mužskou endokrinní funkci – produkují androgeny a antimülleriánský hormon (AMH – anti-Müllerian hormone). Vzhledem k tomu, že organizmu chybějí funkční androgenní receptory, dochází k poruše vývoje vnitřního i zevního mužského genitálu. Dochází k agenezi nadvarlat, ductus deferens (závisí na testosteronu), dále semenných váčků, prostaty, penisu a skrota (závisí na dihydrotestosteronu). V neposlední řadě je těžce narušena spermatogeneze. Dihydrotestosteron vzniká konverzí z testosteronu a je také zodpovědný za sestup varlat. Ektopická varlata pak mohou být lokalizována v pánvi, tříselném kanálu či ve velkých stydkých pyscích. AMH produkovaný ze Sertoliho-Leydigových buněk varlete inhibuje vývoj vejcovodů , dělohy a proximální části pochvy z paramezonefrických vývodů . Estrogeny, které se syntetizují v testes a také aromatizací testosteronu v periferních tkáních, jsou následně v pubertě zodpovědné za růst prsů (telarché). V důsledku androgenní insenzitivity chybí axilární ochlupení, pubické je velmi sporé (tzv. hairless woman). Pacientky s CAIS se cítí ženami, vypadají jako ženy, jsou zpravidla vyššího vzrůstu, atraktivního vzhledu, s vyvinutými prsy, nejsou ale fertilní. Dysgenetické gonády přinášejí riziko malignizace. To se pohybuje v dětství mezi 2 a 5 %, v pubertě stoupá na 20 %. Je tedy indikována gonadektomie a hormonální substituční terapie.
Kazuistika
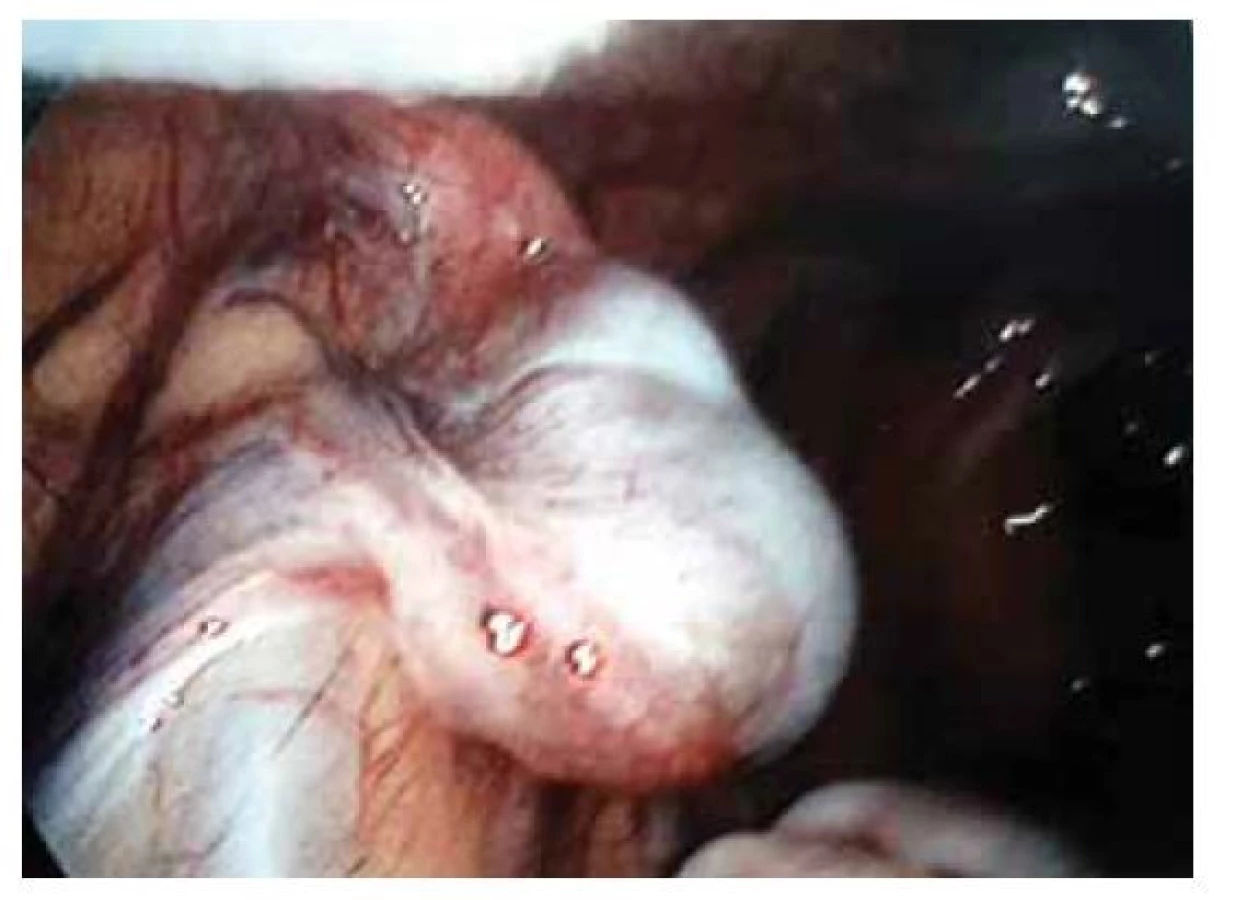
Pacientka (17 let) přichází do gynekologické ambulance pro primární amenoreu. Po odebrání anamnézy, zhodnocení vývoje sekundárních pohlavních znaků a po komplexním gynekologickém a ultrazvukovém vyšetření je vysloveno podezření na agenezi dělohy. Zevní genitál je normálního vzhledu, sekundární pohlavní znaky jsou vyvinuty, pochva je nezkrácena, ale slepě ukončena, čípek nedetekován. Při transvaginální sonografii se děloha nezobrazuje, pravé ovarium je bez folikulů, levé ovarium s dobře ohraničeným hypoechogenním solidním tumorem charakteru fibromu, velikosti 40 x 30 mm, dopplerovské vyšetření s hraniční perfuzí. Bez nálezu další patologie v malé pánvi. Laboratorní vyšetření jsou v normě, vč. onkomarkerů (hCG, AFP, CEA, CA-125). Následně je indikována diagnostická laparoskopie, která potvrzuje agenezi dělohy (obr. 1). V laparoskopickém obraze dominují bilaterálně nepřímé inguinální hernie (obr. 2). Pravý vaječník vč. vejcovodu je hypoplastický, nejeví známky ovulace (obr. 3). Levý vaječník má velikost 5 cm a vejcovod nedetekujeme (obr. 4). Při exploraci laparoskopickými nástroji jsou verifikovány tři tuhé tumory charakteru fibromu (3, 2,5 a 1,5 cm), které jsou bez porušení pouzdra exstirpovány a extrahovány z dutiny břišní. Ostatní orgány a povrchy dutiny břišní jsou bez patologie. Laparoskopický výkon je veden tak, aby bylo zachováno maximum zárodečné tkáně. Histologické vyšetření prokazuje dobře diferencovaný tumor ze Sertoliho-Ley-fardigových buněk. Následně je doplněno CT vyšetření břicha a pánve, které kromě ageneze dělohy a špatně přehledných hypoplastických ovarií nepotvrzuje žádnou patologii, lymfadenopatie není přítomna. Pacientka je dále referována ke konzultaci na dětskou onkologii a na genetické vyšetření. Histologicky se jedná o dobře diferencovaný gonadostromální tumor ovaria ze Sertoliho-Leydigových buněk, hodnota Ki67 je 2 %. Tumor je zařazen do klinického stadia IA dle FIGO, dle aktuálních ESGO-SIOPe guidelines není indikována adjuvantní léčba, je zvolena strategie „wait and watch“. Překvapivý závěr ale přináší genetické vyšetření, které u pacientky prokazuje karyotyp 46,XY. Po došetření genů spojených s gonadální dysgenezí je stanovena konečná diagnóza – syndrom úplné androgenní insenzitivity. Je doporučena oboustranná gonadektomie, hormonální substituce a pravidelné kontroly v onkologické ambulanci. Bilaterální gonadektomii provádíme v druhé době, histologický nález odpovídá obrazu dysgenetické testikulární tkáně, bez známek atypií či neoplastických změn. Pacientka je po operaci zajištěna estrogenní hormonální substitucí.
Fig. 1. Laparoscopic verification of uterine agenesis.
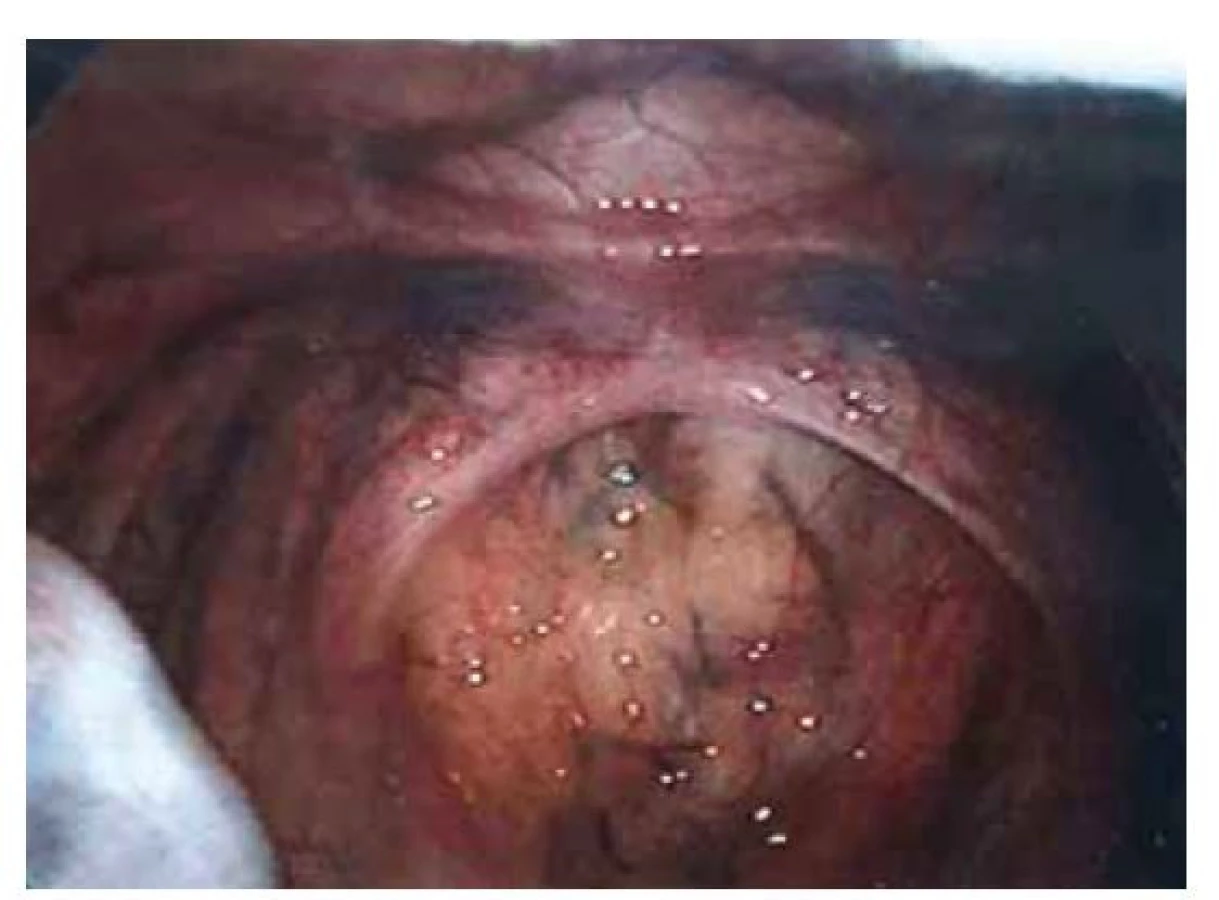
Fig. 2. Indirect inguinal hernia on the left side.
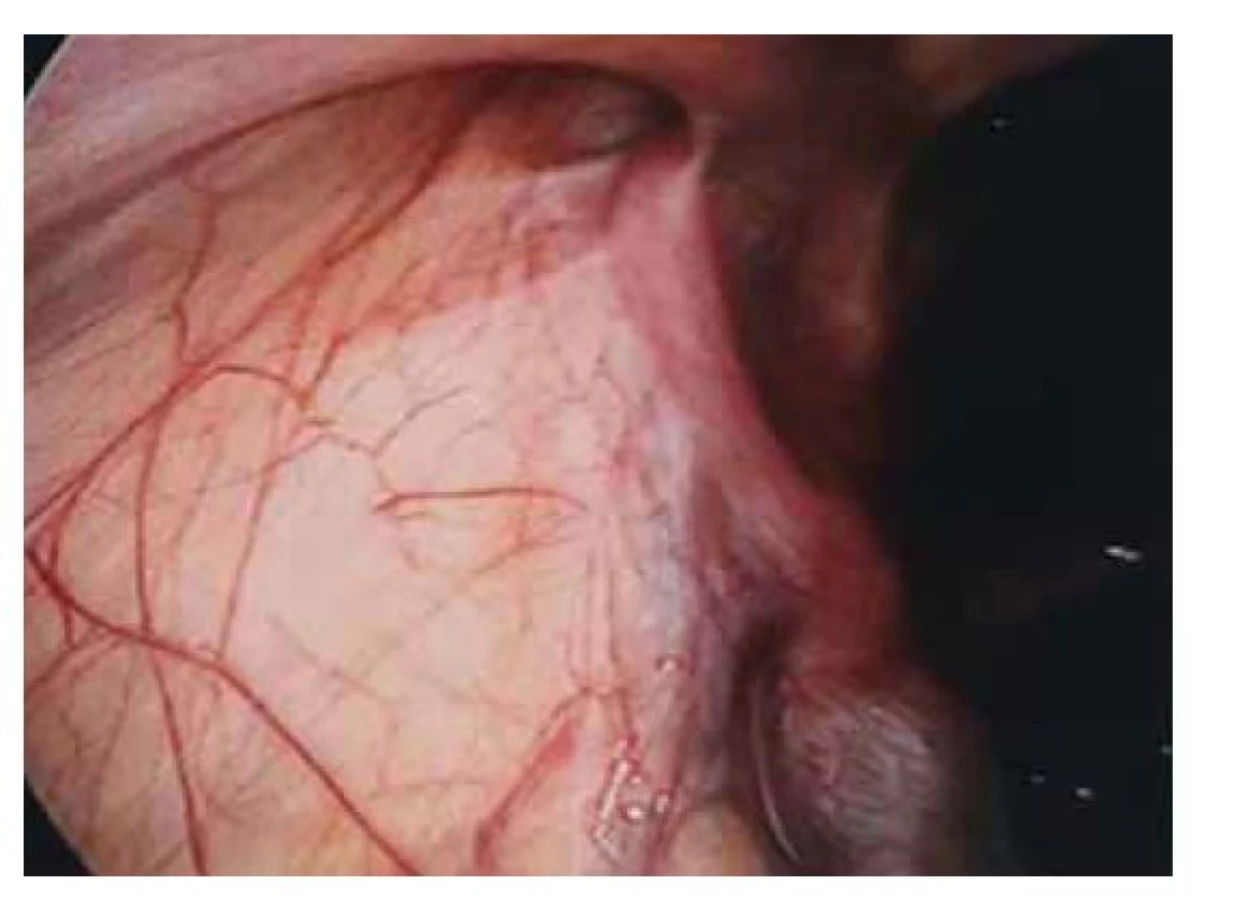
Fig. 3. Hypoplasia of the right adnexa.
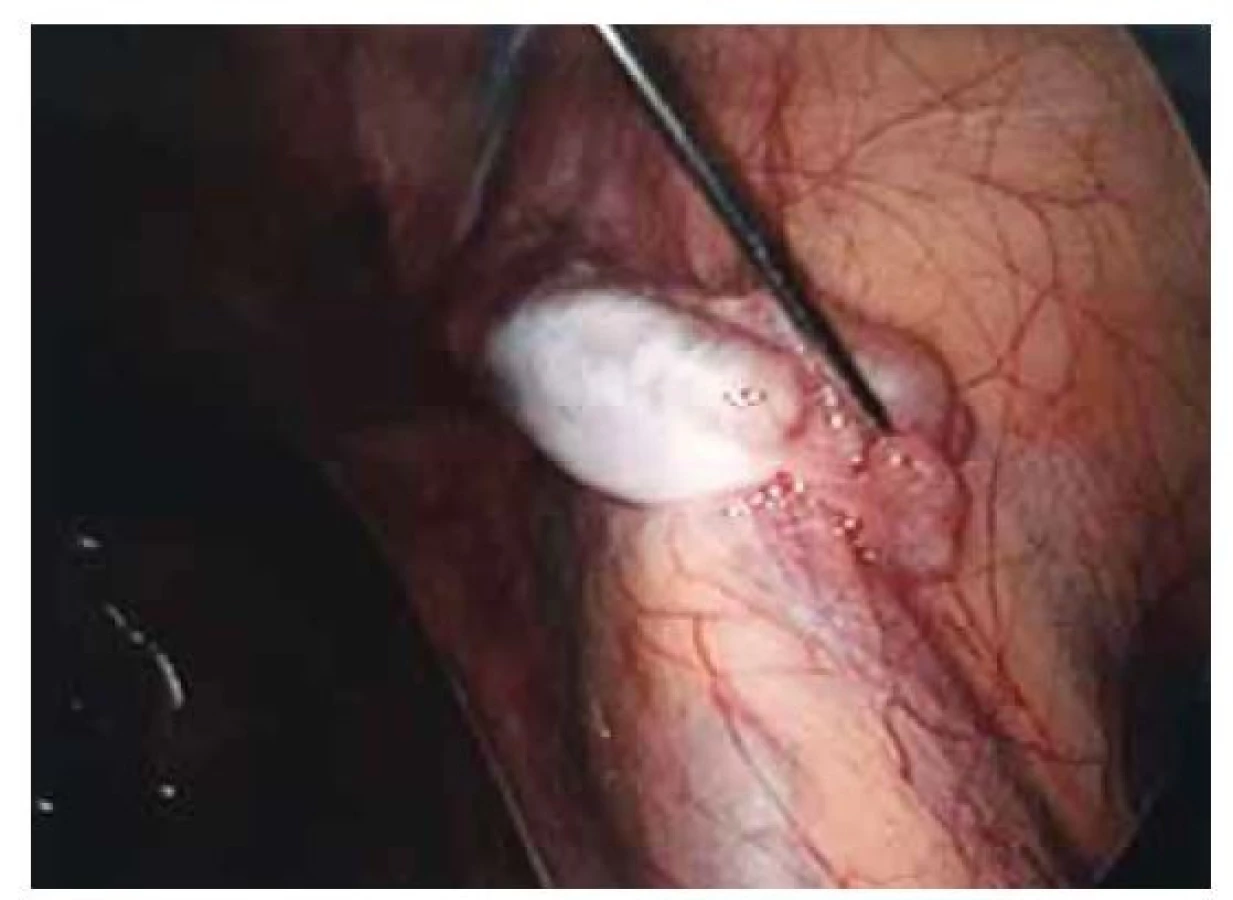
Fig. 4. Left gonad with agenesis of the tube.
Diskuze
V této kazuistice jsme syndrom úplné androgenní insenzitivity diagnostikovali pacientce původně vyšetřované pro primární amenoreu. Dalšími indiciemi, které mohou vést k této diagnóze, jsou nemožnost pohlavního styku, tříselná kýla u holčičky či mladé dívky, anamnéza onemocnění v rodině či diskrepance karyotypu amniocentézy a fenotypu na ultrazvukovém screeningu či po porodu.
V rámci diferenciální diagnostiky ageneze dělohy přichází v úvahu Mayer- -Rokitansky-Küster-Hauserův syndrom (MRKH). Je definován jako vrozená ageneze dělohy a horních dvou třetin pochvy u žen s ženským karyotypem (46,XX). Vaječníky a vejcovody jsou vyvinuty, a proto mají ženy s MRKH syndromem normální ženské sekundární pohlavní znaky. Na rozdíl od pacientek s CAIS je ale pochva u těchto pacientek významně zkrácena (vaginální rudiment délky pouze 1–2 cm), případně je přítomná pouze tzv. vaginální jamka (vaginal dimple) [4].
Náhodným nálezem u naší pacientky byl tumor dysgenetického varlete. V literatuře se popisuje různé riziko malignizace dysgenetických gonád, procentuálně se pohybuje mezi 0,8 a 22 %, zvyšuje se v pubertě [5]. Proto je doporučena oboustranná gonadektomie. Názory na načasování operace se ale různí. Část autorů se přiklání k časné gonadektomii, ideálně ihned po odhalení diagnózy. Puberta se poté navozuje farmakologicky. Druhá skupina zastává přístup postpubertální gonadektomie pro možnost spontánního nástupu a vývoje sekundárních pohlavních znaků při relativně nízkém riziku malignity [6]. Definitivní rozhodnutí o postupu by mělo následovat po pečlivém poučení pacientky a její rodiny.
Nejčastějšími typy nádorů dysgenetických gonád jsou gonadoblastom, dysgerminom/ seminom a embryonální karcinom. Méně často se objevují smíšené germinální nádory, nádor ze žloutkového váčku, choriokarcinom či nádory ze Sertoliho-Leydigových buněk [7]. U naší pacientky byl histologicky potvrzen dobře diferencovaný tumor ze Sertoliho-Leydigových buněk. Jedná se o vzácný gonadostromální nádor, který obsahuje Sertoliho a Leydigovy buňky, což jsou nezárodečné podpůrné buňky mužských gonád. Vzniká z primitivních buněk pregranulózy a jeho výskyt je vázán na dysgenezi gonád, syndrom DICER1 nebo Peutz-Jeghersův syndrom. Podle WHO klasifi kace jsou tyto nádory děleny na dobře, středně a špatně diferencované a retiformní. Stupeň diferenciace koreluje s prognózou. Dobře diferencované nádory se obvykle chovají jako benigní, zatímco špatně diferencované mají maligní povahu [8].
Další problematikou, kterou se u pacientek s CAIS zabýváme, je hormonální substituce. Ta je nezbytná po proběhlé gonadektomii k navození vývoje sekundárních pohlavních znaků, k prevenci osteoporózy, kardiovaskulárních onemocnění a k podpoře celkového prospívání a sexuálních funkcí. Standardně se preferuje kontinuální užívání estrogenů, ale v zahraniční literatuře jsou uváděny i zkušenosti s podáváním androgenů. Testosteron je pacientkami dobře tolerován a je stejně bezpečný jako estrogeny. Dle studií je vhodnější zejména u pacientek se sníženou sexuální funkcí a nižším libidem [9]. Gestageny vzhledem k absenci dělohy nejsou indikovány.
Jak již bylo výše zmíněno, některé pacientky s CAIS se potýkají s problémem dyspareunie. Ta vzniká na podkladě nedostatečně prostorné pochvy. V těchto případech je potřebná úprava délky pochvy, vaginální dilatace či vaginoplastika. Vaginální dilatace různými dilatátory či fantomy je většinou postačující. Pokud tato metoda selže, byly navrženy různé operační postupy a techniky léčby slepě ukončené pochvy, které zahrnují vaginoplastiku za využití různých autologních štěpů (bukální sliznice, peritoneum, část tenkého či tlustého střeva, kožní laloky) anebo balonkovou vaginoplastiku [1,10].
Závěr
Syndrom úplné androgenní insenzitivity je vzácné genetické onemocnění projevující se různým stupněm feminizace u jedinců s mužským karyotypem. Mělo by se na ni pomýšlet zejména při diferenciálně diagnostické rozvaze primární amenorey. Zvlášť při podezření na agenezi dělohy by mělo vždy následovat genetické vyšetření karyotypu. Pacientky s tímto onemocněním vypadají jako ženy, cítí se ženami, jsou ale infertilní. Kauzální léčba neexistuje. Komunikace s pacientkou i její rodinou vyžaduje citlivý přístup, součástí péče by měla být psychologická podpora a mezioborová spolupráce gynekologa, pediatra, endokrinologa a genetika.
ORCID autorů
V. Gamcová 0000-0002-2991-0628
J. Eim 0000-0002-9168-5249
I. Meixnerová 0000-0002-4837-4795
R. Hudeček 0000-0003-0617-0126
Doručeno/ Submitted: 6. 2. 2022
Přijato/ Accepted: 20. 2. 2022
MUDr. Viktória Gamcová
Gynekologicko-porodnické oddělení
Nemocnice Vyškov, p.o.
Purkyňova 36
682 01 Vyškov
Zdroje
1. Gottlieb B, Trifiro MA. Androgen insensitivity syndrome. GeneReviews, 2017 [online]. Available from: https:/ / www.ncbi.nlm.nih. gov/ books/ NBK1429/ .
2. Khollová S, Hrdonková E, Pomahačová R. Syndrom ú plné androgenní insenzitivity – kazuistika. Ceska Gynekol 2014; 79(1): 38–42.
3. Boehmer AL, Brinkmann O, Brü ggenwirth H et al. Genotype versus phenotype in families with androgen insensitivity syndrome. J Clin Endocrinol Metab 2001; 86(9): 4151–4160. doi: 10.1210/ jcem.86.9.7825.
4. Chmel R Jr, Pastor Z, Mužík M et al. Syndrom Mayer-Rokitansky-Küster-Hauser – agene ze dělohy a pochvy: aktuální znalosti a terapeutické možnosti. Ceska Gynekol 2019; 84(5): 386–392.
5. Looijenga LH, Hersmus R, Oosterhuis JW et al. Tumor risk in disorders of sex development (DSD). Best Pract Res Clin Endocrinol Metab 2007; 21(3): 480–495.
6. Batista RL, Costa EM, de Santi Rodrigues A et al. Androgen insensitivity syndrome: a review. Arch Endocrinol Metab 2018; 62(2): 227–235. doi: 10.20945/ 2359-3997000000031.
7. Patel V, Casey RK, Gomez-Lobo V. Timing of gonadectomy in patients with complete androgen insensitivity syndrome-current recommendations and future directions. J Pediatr Adolesc Gynecol 2016; 29(4): 320–325. doi: 10.1016/ j. jpag.2015.03.01.
8. Plevova P, Gerzova H. Vzácné pediatrické ovariální tumory a jejich genetické příčiny. Klin Onkol 2019; 32 (Suppl 2): S79–S91. doi: 10.14735/ amko2019S79.
9. Birnbaum W, Marshall L, Werner R et al. Oestrogen versus androgen in hormone-replacement therapy for complete androgen insensitivity syndrome: a multicentre, randomised, double-dummy, double-blind crossover trial. Lancet Diabetes Endocrinol 2018; 6(10): 771–780. doi: 10.1016/ S2213-8587(18)30 197-9.
10. Hořejší J, Kosová H a kol. Dětská gynekologie. 2. vyd. Praha: Mladá fronta 2019: 243–245.
Štítky
Dětská gynekologie Gynekologie a porodnictví Reprodukční medicínaČlánek vyšel v časopise
Česká gynekologie

2022 Číslo 3
Nejčtenější v tomto čísle
- Gravidita v jizvě po císařském řezu
- Hydronefróza jako příznak asymptomatické ureterální endometriózy
- Nádory ovaria a genetická dispozice
- Syndrom úplné androgenní insenzitivity – raritní kazuistika malignizace dysgenetických gonád