
Hyperechogenita intestina jako marker cystické fibrózy u plodu
Hyperechogenic fetal bowel as a markerof fetal cystic fibrosis
Introduction:
Hyperechogenic bowel (HB) occurs in 0.1 to 1.8% of normal pregnancies. In most cases it has no consequence for the foetus, but can be associated with cystic fibrosis (CF), chromosomal defects, genetic syndromes, viral infections, gastrointestinal pathology, missed gravidity, IUGR and preterm labour.
Objectives:
Assessment the risk of the foetus having CF or other abnormalities when HB was detected during ultrasound screening in the second trimester of pregnancy in our centre.
Design:
Retrospective study.
Setting:
Department of Obstetrics and Gynecology, Centre of Fetal Medicine and Genetics, KNTB a.s. Zlín.
Methods:
Retrospective analysis of 149 cases of HB between 17 to 22 weeks of pregnancy detected from January 2008 to April 2012.
HB was evaluated according to its degree of echogenicity (Slotnik/Abuhamed classification), presence or absence of other ultrasound markers and the result of first trimester combined screening result. When stage II or III HB and/or borderline risk in first trimester screening, and presence of other ultrasound markers was detected, amniocentesis (AMC) was performed to investigate the karyotype, mutations in the CFTR gene and presence of viral infections (cytomegalovirus and parvovirus B19). If stage I or II HB and/or negative I. trimester screening and no other ultrasound markers, viral infections and mutations in the CFTR gene were investigated form maternal blood. If positive, paternal blood sampling testing for mutation in the CFTR gene was performed. If a mutation was detected in both parents, AMC was performed.
Mutations of the CFTR gene was investigated with a com-mercial panel of 33 to 50 most common mutations.
Postnatally the outcome of neonatal screening for CF(IRT) and any newborns with congenital malformations were ascertained.
Results:
HB was seen in 149 foetuses, AMC was performed in 94 (63%), and blood sampling in 55 (37%). Two mutations in the CFTR gene associated with a severe form of CF (deltaF508/3849 KBC +10 T) were found in one foetus from the AMC group with stage III HB. The parents decided to terminate the pregnancy.
The incidence of HB in our group was 0.7%. In 4 foetuses (2.7%) with stage II HB heterozygous deltaF508 mutation was found, in the rest no mutations were detected. Parents of heterozygous carriers underwent genetic consultation. Postnatal CF screening (IRT level from a heel prick sample) was negative; therefore no further molecular genetic analysis was performed.
Infection was detected in three foetuses; one case was managed with intrauterine transfusion and in the other two cases parents decided for termination. Four cases (2.7%) were terminated because of severe congenital anomalies. Minor congenital abnormalities were detected in seven (4.7%) cases. Intrauterine death was detected in three (2%) pregnancies.
Conclusion:
Based on our results, HB can be considered as a significant marker for the risk of CF, especially in HB stages II and III. It also demonstrates the importance of this marker for the risk of other foetal abnormalities.
Keywords:
hyperechogenic bowel, cystic fibrosis, mutation, amniocentesis, viral infection, chromosomal abnormalities
Authors:
M. Sukupová 1,2; I. Dhaifalah 2,3; Z. Adamík 1; J. Havalová 1,2
Authors‘ workplace:
Gynekologicko-porodnické oddělení, Krajská nemocnice T. Bati, a. s. Zlín, přednosta MUDr. Z. Adamík, Ph. D.
1; Centrum fetální medicíny a lékařské genetiky, Krajská nemocnice T. Bati, a. s. Zlín
vedoucí pracoviště doc. MUDr. I. Dhaifalah, Ph. D.
2; Ústav lékařské genetiky LF UP, Olomouc, přednostka doc. MUDr. I. Dhaifalah, Ph. D.
3
Published in:
Ceska Gynekol 2015; 80(1): 20-24
Overview
Úvod:
Hyperechogenita intestina (HI) se vyskytuje u 0,1–1,8 % normálních těhotenství. Lze ji považovat za normální nález, ale může být markerem cystické fibrózy chromozomálních vad, genetických syndromů a virových infekcí. Může souviset také s patologií GIT, existuje riziko missed gravidity, IUGR a předčasného porodu.
Cíl studie:
Stanovení rizika postižení plodu cystickou fibrózou (CF) při nálezu hyperechogenity intestina během ultrazvukového vyšetření ve II. trimestru gravidity. Stanovení rizika také pro další patologie plodu při nálezu HI.
Typ studie:
Retrospektivní studie.
Název a sídlo pracoviště:
Gynekologicko-porodnické odd., Centrum fetální medicíny a lékařské genetiky, Krajská nemocnice T. Bati, a.s., Zlín.
Metody:
Metodou je retrospektivní analýza zahrnující 149 případů nálezu HI při UZ vyšetření mezi 17.–22. t.g. během období 1/2008–4/2012.
Při nálezu HI byl zhodnocen její stupeň podle Slotnika/Abuhamada, byl zhodnocen výsledek I. trimestrálního screeningu a byla zhodnocena přítomnost dalších ultrazvukových markerů. Při HI 2. nebo 3. stupně a/nebo při současném hraničním riziku v I. trimestrálním screeningu, stejně tak při dalších UZ markerech, byla indikována AMC k vyšetření karyotypu, mutací v genu CFTR a virových infekcí (CMV a PV B19). Při HI 1. nebo 2. st. a/nebo negativním I. trimestrálním screeningu a absenci dalších UZ markerů, byla nejprve odebrána krev matky (PK) k vyšetření virových infekcí a mutací v genu CFTR, při pozitivním nálezu byla vyšetřena i krev otce. Při diagnostikované mutaci v genu CFTR u rodičů byla indikována AMC.
Analýza mutací v genu CFTR zahrnovala vyšetření komerčního panelu 33–50 nejčastějších mutací.
Následně byl zjištěn outcome u novorozence po porodu – výsledek novorozeneckého screeningu na CF (IRT) a případné VVV u novorozence.
Výsledky:
Ze souboru 149 pacientek s nálezem HI bylo provedeno 94 (63 %) AMC a 55 (37 %) odběrů PK. V souboru AMC byl zachycen 1 plod s HI 3. st. se 2 mutacemi v genu CFTR podmiňujícími závažnou formu CF (deltaF508/3849+10kbC T), rodiče se rozhodli ukončit graviditu. Incidence tohoto nálezu v našem souboru byla 0,7 %. U 4 (2,7 %) plodů s HI 2. st. byla nalezena 1 mutace – deltaF508 v heterozygotním stavu (plod přenašeč). Rodiče absolvovali genetickou konzultaci. Po porodu byl vyšetřen pouze IRT (v normě), proto další analýza mutací genu v CFTR nebyla provedena. U ostatních plodů nebyla zjištěna žádná mutace v genu CFTR.
Infekce byla zachycena u tří plodů, v jednom případě byly indikovány intrauterinní transfuze, ve dvou případech se rodiče rozhodli k ITP. U čtyř případů (2,7 %) byly zachyceny závažné VVV a rodiče se rozhodli k ukončení gravidity. Sedm případů (4,7 %) VVV bylo méně závažných. U tří případů (2 %) došlo k missed graviditě.
Závěr:
Na základě našich výsledků lze HI považovat za signifikantní marker pro riziko postižení plodu CF, především u HI 2.–3. stupně. Naše výsledky také dokazují důležitost tohoto markeru i pro jiné, výše uvedené patologie.
Klíčová slova:
hyperechogenita intestina, cystická fibróza, mutace, amniocentéza, virové infekce, chromozomální vady
ÚVOD
Hyperechogenita intestina (HI) se vyskytuje u 0,1–1,8 % normálních těhotenství [8, 12]. Lze ji považovat za normální nález, ale může být markerem cystické fibrózy (CF) [1, 3, 7, 8, 12–15], chromozomálních vad, genetických syndromů [1, 4, 8, 12–15], virových infekcí [1, 4, 8, 12, 14, 15], může souviset s patologií GIT [1, 4, 8, 10, 12, 14], existuje riziko missed gravidity [1, 4, 7, 10, 11, 12, 14], IUGR [4, 11, 12, 14, 15] a předčasného porodu [12, 14].
CF je nejčastější metabolické onemocnění podmíněné AR dědičností, s incidencí 1:2700 živě narozených dětí [17], incidence přenašečů v populaci je 1:25 [16]. Je to onemocnění multiorgánové, postihující plíce, pankreas, trávicí ústrojí a reprodukční orgány. Onemocnění je způsobeno mutací v genu CFTR. Doposud bylo identifikováno více než 1700 mutací v genu CFTR [2 ,3]. CFTR gen je lokalizován na chromozomu 7 (7q31), je 250 kb dlouhý a má 27 exonů [2, 19]. Gen CFTR kóduje CFTR protein (cystic fibrosis transmembrane conductance regulator protein) [3], který ovlivňuje transport iontů chloru, sodíku a vody přes apikální membránu specializovaných epiteliálních buněk. Mutace v genu CFTR způsobí, že CFTR protein je defektní, je redukován nebo chybí. Důsledkem je defekt transportu iontů a hromadění hlenu na apikální membráně buněk, což je patofyziologickým podkladem CF [9]. Rozdílné mutace způsobují rozdílné postižení CFTR proteinu, a tím i různou závažnost klinických příznaků [2].
CÍLE
Stanovení rizika postižení plodu cystickou fibrózou (CF) při nálezu hyperechogenního intestina (HI) během ultrazvukového vyšetření ve II. trimestru gravidity. Stanovení souvislosti mezi mírou postižení plodu CF a stupněm hyperechogenity intestina. Stanovení rizika pro další patologie u plodu při nálezu HI.
METODY
Metodou je retrospektivní analýza zahrnující 149 případů nálezu hyperechogenního intestina (HI) u plodu během ultrazvukového vyšetření mezi 17. a 22. t.g. během období 1/2008–4/2012.
Při nálezu hyperechogenity intestina byl zhodnocen její stupeň podle Slotnika/Abuhamada (srovnání echogenity intestina s echogenitou ilických kostí) [13], byl zhodnocen výsledek I. trimestrálního screeningu a byla zhodnocena přítomnost dalších ultrazvukových markerů ve II. trimestru gravidity. Při nálezu hyperechogenity středně těžké nebo závažné (2.–3. stupně), a/nebo při současném hraničním riziku v I. trimestrálním screeningu, stejně tak při dalších UZ markerech, byla rodičům nabídnuta AMC k vyšetření karyotypu plodu, vyšetření mutací v genu CFTR a vyšetření virových infekcí (CMV a PV B19). Při nálezu hyper-echogenity mírné (1. st.) nebo středně závažné (2. st.) a/nebo negativním I. trimestrálním screeningu a absenci dalších UZ markerů, byla nejprve odebrána krev matky k vyšetření mutací v genu CFTR, při nálezu mutace u matky byla vyšetřena na mutace v genu CFTR i krev otce. Při pozitivním nálezu mutací v genu CFTR z krve rodičů byla opět nabídnuta AMC k vyšetření mutací v genu CFTR u plodu (zároveň byl taktéž vyšetřen karyotyp plodu a virové infekce).
Analýza mutací v genu CFTR zahrnovala vyšetření komerčního panelu 33/39/50 (počet vyšetřovaných mutací se liší podle roku vyšetření) nejčastějších mutací v naší populaci metodou PCR. Nejčastější mutace: delF508 – 70 % všech mutací [8], CFTRdele2,3, G542X, G551D, R553X, N1303K [7, 9, 17]. Vyšetření těchto mutací dokáže zachytit 92–93 % popsaných mutací v genu CFTR (Kit F EU 2 firmy Elucigene). Dále byly vyšetřeny polytymidinové alelické varianty v intronu 8 (IVS8polyT) a počet TG replikací. Varianta polytymidinového traktu existuje ve třech běžných alelách T(5), T(7), T(9). Číslo 5, 7 a 9 značí počet tymidinů. Čím je počet tymidinů nižší, tím spíše dochází k sestřihu exonu 9 genu CFTR s menší účinností. Tato účinnost je dále ovlivněna počtem přilehlých (TG) repetic: čím je vyšší počet TG repetic, tím je nižší účinnost sestřihu. Při sestřihu exonu 9 s nižší účinností dochází ke vzniku menšího množství funkčního produktu genu CFTR – CFTR proteinu [2, 19]. Při rozšířené mutační analýze se detekují mutace v 19 oblastech genu CFTR (exon 2, intron 1/intron 3, exon 3, exon/intron 4…), pokud ani toto neodhalí patologii v genu CFTR, provádí se v indikovaných případech sekvence celého genu CFTR [19].
Z virových infekcí byly standardně vyšetřeny DNA CMV a PV B19 metodou PCR. Karyotyp byl vyšetřen metodou PCR na trizomii 21, 18, 13, dále pokračovala klasická cytogenetická analýza k vyšetření kompletního karyotypu. V indikovaných případech byly vyšetřeny genetické syndromy metodou microarray.
Při pozitivním nálezu jedné mutace u plodu byl plod označen jako přenašeč a rodičům byla nabídnuta genetická konzultace. Zde bylo také zhodnoceno reziduální riziko přítomnosti druhé, vzácnější, standardně nevyšetřované mutace a případně bylo indikováno další vyšetření. V případě nálezu dvou mutací podmiňujících závažnou formu CF měli rodiče možnost se po absolvování genetické konzultace rozhodnout o ukončení gravidity podle zákona.
Následně byl po porodu zjištěn outcome u novorozence: výsledek novorozeneckého screeningu na CF (výsledek IRT) a výsledek potního testu, dále byl zjišťován zdravotní stav a případné VVV u novorozence.
Novorozenecký screening, tzv. IRT test, se v ČR provádí od 1. 10. 2009. Provádí se ze suché kapky krve novorozence zároveň se screeningem na ostatní metabolické vady. Vyšetřuje se hladina imunoreaktivního trypsinogenu (IRT) imunoanalytickou metodou. Je to produkt slinivky břišní a jeho hladina bývá v krvi dětí nemocných CF zvýšená [18]. Ovšem ne všechny děti s vysokým IRT mají CF [16]. Pokud je IRT vyšší, je indikováno vyšetření mutací v genu CFTR. IRT ovšem neodhalí heterozygoty mutací v genu CFTR, tzn. nosiče CF, jelikož není ovlivněna tvorba trypsinogenu. Může být také falešně negativní u dětí s mekoniovým ileem. V případě nálezu jedné nebo dvou mutací v genu CFTR je indikováno provedení potního testu. Ten vyloučí případnou záměnu vzorku a potvrdí diagnózu. U dětí s nalezenou jednou mutací odliší zdravé nositele a děti nemocné CF, kde je potřeba pátrat po druhé, vzácnější mutaci. Stupeň IRT taktéž neodráží závažnost onemocnění CF [18].
VÝSLEDKY
Ze souboru 149 pacientek s nálezem hyper-echogenního intestina u plodu bylo provedeno94 AMC (63 %) a 55 (3 7%) odběrů PK matky k vyšetření mutací v CFTR genu a infekcí (+ 9 vyšetření PK otců). Incidence hyperechogenity intestina byla 1,1 % (tab. 1).
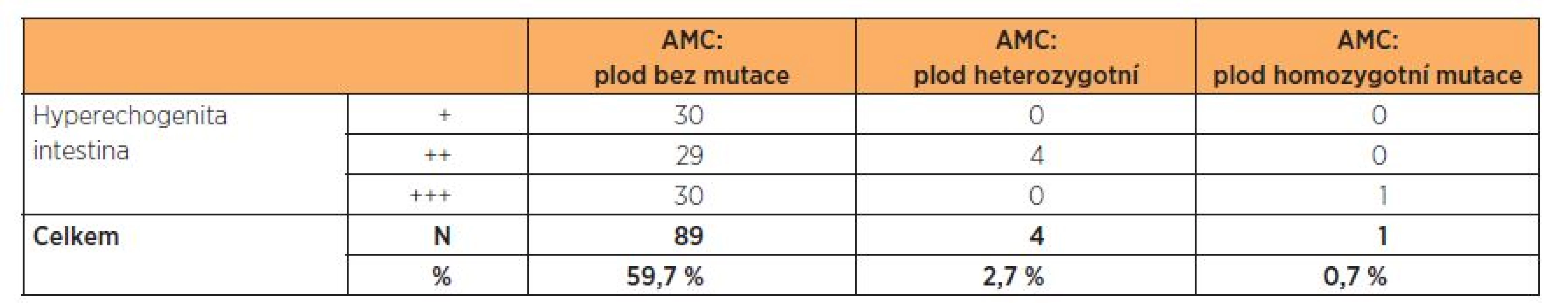
Ze souboru AMC byl zachycen jeden plod s HI 3. st. se dvěma mutacemi v genu CFTR podmiňujícími závažnou formu CF (deltaF508/3849+10kbC T),rodiče se rozhodli ukončit graviditu. Incidence tohoto nálezu byla 1,7 %.
U čtyř plodů s HI 2.–3. st. byla nalezena jedna mutace – deltaF508 v heterozygotním stavu (plod označen jako přenašeč). Incidence přenašečů CF byla 2,7 %. Rodiče absolvovali genetickou konzultaci, rozhodli se pokračovat v graviditě. Po porodu byl u těchto plodů vyšetřen pouze IRT, který byl v normě, proto další analýza mutací genu v CFTR nebyla provedena. Děti jsou nadále sledovány obvodním pediatrem, t.č. jsou bez potíží. Pouze u jednoho dítěte je popisováno vyšší zahlenění, zatím není dáváno do souvislosti s CF. Potní test byl vyšetřen pouze u jednoho dítěte, byl v normě, u dalších dětí zatím proveden nebyl. Vzhledem k výše uvedenému je v plánu další genetická konzultace a dovyšetření dětí po porodu.
U 89, tj. 59,7 %, plodů ze souboru AMC nebyla zjištěna žádná mutace v genu CFTR.
U jednoho plodu s hyperechogenitou 3. st. byla diagnostikována akutní CMV infekce, rodiče se taktéž rozhodli k ITP. U dvou plodů s HI 3. st. jsme diagnostikovali akutní infekci způsobenou PV B19. U prvního plodu ve stadiu závažné anémie a hydropsu plodu se srdečním selháním se rodiče rozhodli podstoupit ITP. U druhého plodu byl záchyt včasný, plodu byly podány opakované intrauterinní transfuze, novorozenec je po porodu bez zdravotních potíží. U tří plodů s hyperechogenitou 2. a 3. st. byla stanovena diagnóza genetického syndromu a rodiče se opět rozhodli k ITP. U jednoho plodu s HI 3. st. byly následně diagnostikovány mikrocystické ledviny s anhydramniem, tento nález byl taktéž indikací k ITP z genetické indikace. U dvou plodů s HI 3. st. nastala missed gravidita do III. trimestru gravidity,u jednoho plodu s HI 3. st. došlo k missed graviditě ve III. trimestru gravidity vlivem strangulace pupečníku. U jednoho plodu s HI 3. st. došlo k PPROM v 26. t.g. s následným partus praematurus (v tomto případě nebyla provedena AMC, byla indikována, ale rodiči neakceptována, proto byl proveden pouze odběr PK) (tab. 2).

V souboru vyšetřených PK jsme zachytili 2 mutace v genu CFTR u matek, tento nález byl následně indikací k vyšetření otce. U 12 matek byly zachyceny polyT varianty v IVS8 s TG repeticemi, bez nálezu mutace v genu CFTR, tyto nálezy byly následně taktéž indikací k vyšetření otce. V obou případech nebyla u otců diagnostikována žádná mutace, rodiče podstoupili genetickou konzultaci se závěrem: minimální riziko a nepravděpodobné postižení plodu CF.
U 39 žen bylo vyšetření mutací v genu CFTR z PK zcela negativní, tzn. nepravděpodobné postižení plodu CF.
V našem souboru plodů s hyperechogenitou intestina vyšetřovaných na CF jsme nezachytili žádnou chromozomální vadu – trizomii 21. To si vysvětlujeme včasným záchytem trizomie 21 již v I. trimestru gravidity (jeden případ trizomie 21 zachycený na základě hyperechogenity intestina v 16. t.g., kdy I. trimestrální screening proveden nebyl, do našeho souboru nebyl zařazen).
ZÁVĚR
Na základě našich výsledků lze hyperechogenitu intestina považovat za signifikantní marker pro riziko postižení plodu CF, především u hyperechogenity 2.–3. st. Incidence závažné formy CF je v našem souboru 1,7 % proti incidenci ve všeobecné populaci 0,032 % [8].
V případě záchytu dvou mutací a rizika závažné formy CF u plodu mají rodiče možnost se včas rozhodnout, zda chtějí graviditu ukončit. V případě záchytu jedné mutace u plodu lze zhodnotit, zda může být spojena se závažnou, či mírnější formou CF, je zhodnoceno riziko pravděpodobného nosičství. Lze, za předpokladu dobré spolupráce s neonatology a obvodními pediatry, tyto rizikové novorozence podrobně vyšetřit po porodu a sledovat. Na základě dostupných údajů je prokázána důležitost včasného záchytu onemocnění CF. Tento včasný záchyt umožňuje včasnou léčbu, eliminuje závažné komplikace a prodlužuje délku života i u mírnějších forem onemocnění.
Naše výsledky také dokazují důležitost tohoto markeru i pro jiné patologie, především infekce plodu, chromozomální vady a genetické syndromy, v neposlední řadě ale i pro možnost IUGR či missed gravidity. Pečlivé sledování těhotných se záchytem hyperechogeního intestina, při vyloučení jiných patologií, tak může do značné míry předejít i těmto závažným stavům, případně je do určité míry pozitivně ovlivnit.
MUDr. Michaela Sukupová
Gynekologicko-porodnické odd.
Centrum fetální medicíny a lékařské genetiky
KNTB a.s.
Havlíčkovo nábř. 600
762 75 Zlín
e-mail: m.sukupova@seznam.cz
Sources
1. Abramowicz, MJ., Dessars, B., Sevens, C., et al. Fetal bowel hyperechogenicity may indicie mild atypical cystic fibrosis: a case associated with a komplex CFTR allele. J Med Genet, 2000, 37 (http://jmedgenet.com/cgi/conent/full/37/8/e15).
2. Balaščáková, M., Piskáčková, T., Holubová, A., a kol. Současné metodické postupy a přehled neimplantační, prenatální a postnatální DNA diagnostiky v České republice. Čes-slov Pediat, 2008, 63, 2, s. 62–75
3. Becdelievre, A., Costa, C., LeFloch, A., et al. Notable contribution of large CFTR gene rearrangements to the diagnosis of cystic fibrosis in fetuses with bowel anomalies. Eur J Hum Genet, 2010, 18, p. 1166–1169.
4. Ghose, I., Mason, GC., Martinez, D., et al. Hyperechogenic fetal bowel: a prospective analysis of sixty consecutive cases. Brit J Obstet Gynecol, 2000, 107, p. 426–429.
5. Jouannic, JM., Gavard, L., Criquat, J., et al. Isolated fetal hyperechogenic bowel associated with intra-uterine parvovirus B19 infection. Fetal Diagnosis Ther, 2005, 20(6), p. 498–500.
6. Markus-Soekarman, D., Offermans, J., Van den Ouwe-land, AM., et al. Hyperechogenic fetal bowel: counseting difficulties. Eur J Med Genet, 2005, 48(4), p. 421–425.
7. Muller, F., Dommergues, M., Simon-Bouy, B., et al. Cystic fibrosis screening: a fetus with hyperechogenic bowel may be the index case. Medical Genet, 1998, 35, p. 657–660.
8. Muller, F., Simon-Bouy, B., Girodon, E., et al. Predicting the risk of cystic fibrosis with abnormal ultrasound signs of fetal bowel. Amer J Medical Genet, 2002, p. 109–115.
9. Rowe, SM., Miller, S., Sorscher, E., et al. Mechanisms of disease cystic fibrosis, review article. New Engl J Med, 2005, 5, p. 1992–2005.
10. Ruiz, MJ., Thatch, KA., Fisher, JC., et al. Neonatal outcomes associated with intestinal abnormalities diagnosed by fetal ultrasound. J Pediatric Surg, 2009, 4, p. 71–75.
11. Sepulveda, W., Nicolaides, P., Mai, AT. Is isolated second-trimester hyperechogenic bowel a predictor of suboptimal fetal growth? Ultrasound Obstet Gynecol, 1996, 7(2), p. 104–107.
12. Simon-Bouy, B., Satre, V., Ferec, C., et al. Hyperechogenic fetal bowel: a large collaborative study of 682 cases. Amer J Med Genet, 2003, 121A, p. 209–213.
13. Slotnik, RN., Abuhamad, AZ. Prognostic implication of fetal echogenic bowel. Lancet, 1996, 1, p. 85–87.
14. Stringer, MD., Thornton, JG., Mason, GC. Hyperechogenic fetal bowel. Archives of disease in childhood. J Brit Paediatric Assoc, 1996, 74, F1–F2.
15. Strocker, AM., Snijders, RJ., Carlson, DE., et al. Fetal echogenic bowel: parameters to be considered in differential diagnosis. Ultrasound Obstet Gynecol, 2000, 16, p. 519–523.
16. Vávrová, V., Bartošová, D., a kol. Cystická fibrosa – Příručka pro nemocné a jejich rodiče, 2. vyd. Professional Publishing, 2009, ISBN 978-80-7431-000-3.
17. Vávrová, V., Zemková, D., Skalická, V., et al. Cystická fibrosa v ČR. Practicus, 2008, 8, s. 17–21.
18. Votava, F., Adam, T., Zeman, J., Macek, M. Novorozenecký screening – http://www.novorozeneckyscreening.cz.
19. http://www.molekulara.cz/co-vysetrujeme/cysticka fibrosa.
Labels
Paediatric gynaecology Gynaecology and obstetrics Reproduction medicineArticle was published in
Czech Gynaecology

2015 Issue 1
Most read in this issue
- 4G/4G polymorfismus genu pro inhibitor aktivátoru plazminogenu 1 (PAI-1) jako nezávislý rizikový faktor placentární insuficience, způsobující u plodu hemodynamickou centralizaci
- Přední poševní plastika v lokální anestezii
- Rizikové faktory poškození svalů pánevního dna v souvislosti s vaginálním porodem
- Užití transuretrální aplikace polyacrylamid hydrogelu (Bulkamidu®) k léčbě recidivující stresové incontinence moči po selhání efektu páskových operací