
Trombotické mikroangiopatie a těhotenství
Thrombotic microangiopathy and pregnancy
Objective: The aim of this study is to draw attention to a nosological unit called thrombotic microangiopathy (TMA). This syndrome represents a serious pathological condition characterized by microangiopathic haemolytic anemia (MAHA), thrombocytopenia and various organ dysfunction. Patients are most often presented with symptoms of the HELLP syndrome but if the clinical picture is not restituted within 48–72 hours after delivery, other TMAs should be considered.
Setting: Department of Obstetrics and Gynecology, 1st Medical Faculty and General Teaching Hospital Prague; Clinic of Nephrology, 1st Medical Faculty and General Teaching Hospital Prague; Department of Obstetrics and Gynecology, Regional Hospital Kolín.
Design: Review article and case reports.
Methods: Review of the literature and description of two cases of TMA.
Conclusion: The authors present a basic overview of the issue of TMA, which requires interdisciplinary cooperation of obstetricians, anesthesiologists, nephrologists and hematologists. In the second part of the article, we present two TMA case reports and finally show the differential diagnostic and therapeutic scheme as agreed by the authorities in the field.
Keywords:
HELLP syndrome – eculizumab – thrombotic microangiopathy – atypical hemolytic uremic syndrome – hemolytic uremic syndrome – thrombotic thrombocytopenic purpura – acute fatty liver of pregnancy
Autoři:
M. Koucký 1; A. Toman 3; R. Ryšavá 2; A. Pařízek 1
Působiště autorů:
Gynekologicko-porodnická klinika 1. LF UK a VFN, Praha, přednosta prof. MUDr. A. Martan, DrSc.
1; Klinika nefrologie 1. LF UK a VFN, Praha, přednosta prof. MUDr. V. Tesař, MBA, DrSc.
2; Gynekologicko-porodnické oddělení Oblastní nemocnice, Kolín, primář MUDr. A. Toman, MBA
3
Vyšlo v časopise:
Ceska Gynekol 2020; 85(1): 18-28
Kategorie:
Přehled a kazuistiky
Souhrn
Cíl studie: Upozornit na nozologickou jednotku, která se nazývá trombotické mikroangiopatie (TMA). Tento syndrom představuje velmi závažný patologický stav charakterizovaný mikroangiopatickou hemolytickou anémií (MAHA), trombocytopenií a dysfunkcí různých orgánů. Nejčastěji se onemocnění prezentuje pod obrazem HELLP syndromu, pokud ale nedochází k restituci jeho obrazu do 48–72 hodin po porodu, je nutné již uvažovat o jiné TMA.
Název a sídlo pracoviště: Gynekologicko-porodnická klinika 1. LF UK a VFN v Praze; Klinika nefrologie 1. LF UK a VFN v Praze; Gynekologicko-porodnické oddělení, Oblastní nemocnice Kolín, a.s.
Typ práce: Přehledový článek a kazuistiky.
Metodika: Přehled údajů z literatury o problematice a popis dvou případů TMA.
Závěr: Autoři předkládají základní přehled problematiky TMA, která vyžaduje mezioborovou spolupráci porodníků, anestesteziologů, nefrologů a hematologů. V druhé části článku uvádíme dvě kazuistky TMA a nakonec ukazujeme základní diferenciálně diagnostické a terapeutické schéma, tak jak se na něm shodují autority v oboru.
Klíčová slova:
trombotické mikroangiopatie – HELLP syndrom – atypický hemolyticko-uremický syndrom – hemolyticko-uremický syndrom – trombotická trombocytopenická purpura – akutní těhotenská steatóza jater – eculizumab
ÚVOD
Pojem trombotické mikroangiopatie (TMA) představuje velmi závažný patologický stav, kdy dochází k tvorbě trombóz na úrovni kapilár i arteriol v důsledku poškození endotelu. Je provázen mikroangiopatickou hemolytickou anémií (MAHA), trombocytopenií a dysfunkcí různých orgánů, relativně často je navíc spojen s i koagulopatií. Trombotické mikroangiopatie zahrnují velmi nesourodou skupinu syndromů a stavů, kdy ke konečné diagnóze docházíme postupným vylučováním jednotlivých příčin (per exclusionem). V praxi se nejčastěji setkáváme s tím, že těhotné/rodičky/nedělky se prezentují pod obrazem preeklampsie/HELLP syndromu (hemolysis, elevated liver enzymes, low platelets), který zahrnuje obraz MAHA (dynamické snižování hladiny hemoglobinu, zvyšování hladiny bilirubinu, snížení haptoglobinu, přítomnost schistocytů), periportální ischemie jater (elevace transamináz) a trombocytopenie v důsledku vyšší agregace trombocytů v poškozené periferní mikrocirkulaci. HELLP syndrom se také řadí mezi TMA, ale měl by spontánně odeznívat přibližně do 48 až 72 hodin po porodu. Pokud se tak nestane, je velmi důležité pomýšlet na jiné příčiny TMA, které často představují ještě vážnější ohrožení života než HELLP syndrom. Mezi odbornou veřejností v našem oboru je povědomí o této oblasti stále velmi malé. V následujících kapitolách podáme přehled znalostí o současné problematice s tím, že je nutné vyzdvihnout význam diferenciálně diagnostického přístupu; následují dvě kazuistiky pacientek s TMA.
TROMBOTICKÉ MIKROANGIOPATIE – DĚLENÍ
- Trombotická trombocytopenická purpura (TTP) – vrozená a získaná forma
- Hemolyticko-uremický syndrom (HUS), nejčastěji shiga toxin-producing Escherichia coli (STEC-HUS)
- Atypický hemolyticko-uremický syndrom (aHUS) – „complement mediated HUS“
- Těhotenstvím navozené TMA – preeklampsie, HELLP syndrom, akutní těhotenská steatóza jater (acute fatty liver of pregnancy – AFLP)
- Léky navozené TMA (cyklosporin A, takrolimus, klopidrogel, chinin, některá cytostatika)
- TMA po transplantacích plic, ledvin, kostní dřeně
- Diseminovaná intravaskulární koagulopatie
- Pokročilé malignity
- Těžké infekce, sepse
- Těžká systémová či plicní hypertenze
OBECNÉ RYSY PATOFYZIOLOGIE TMA
TMA mají různé spouštění příčiny, ale obecně lze říci, že k jejich konečnému klinickému obrazu vedou dvě základní cesty. První podskupina startujících faktorů vedoucích k poškození endotelu zahrnuje bakteriální endotoxiny (shiga-toxin), léky, viry, druhá pak endogenní – vrozené – faktory. Endogenní faktory, které mohou vést k TMA, jsou ještě poměrně málo prozkoumané a stále přibývají nové informace. Endotelovou dysfunkci může způsobit dysregulace komplementu (deficit regulátorů, nadbytek aktivátorů – bude uvedeno dále), koagulace (abnormality von Wilebrandtova faktoru – vWF), porucha beta oxidace mastných kyselin a působení autoprotilátek. V dalším textu se pokusím shrnout dosavadní poznatky o patofyziologii jednotlivých uváděných TMA. Vzhledem k velké variabilitě exo- či endogenních příčin lze říci, že i dynamika klinického obrazu jednotlivých TMA se může významně lišit a o to větší jsou pak diagnostické rozpaky. Ať už působí jakékoli vlivy, v důsledku se rozvíjí endotelová dysfunkce a snížená tromborezistence. Narušení endotelu s sebou nese vyšší agregaci trombocytů vedoucí k trombocytopenii v krevním obrazu. V mikrocirkulaci se rozvíjí mikroangiopatická hemolytická anémie (MAHA) charakterizovaná přítomností schistocytů. Jedná se o cirkulující fragmenty červených krvinek, které vznikají mechanickým poškozením v krevním oběhu a jsou detekovatelné při posuzování periferního nátěru krve pod mikroskopem. Poškození endotelu, spojené s agregací trombocytů na něm, naplňuje podstatu mikroangiopatie, kdy tento proces v důsledku vede v ischemii a orgánovou dysfunkci. Nejvíce je patrné narušení funkce ledvin, jater a také nespecifické projevy poškození mikrocirkulace mozku, encefalopatie [17].
KLINICKÝ OBRAZ TMA V TĚHOTENSTVÍ A PO PORODU
Klinický obraz TMA je velmi různorodý a jednotlivé nozologické jednotky se při první prezentaci stavu dají od sebe velmi obtížně odlišit. V naprosté většině případů je pracovní diagnóza stanovena jako HELLP syndrom. Klinické projevy jsou často velmi nespecifické. Mezi nejvýznamnější patří „flu-like“ příznaky připomínající virózu, tj. (sub)febrilie, bolesti hlavy, malátnost, únava, nauzea/zvracení, bolesti v podžebří, dalšími příznaky jsou žloutenka, anémie, relativně často je přítomna hypertenze. Bohužel, nezřídka je jako první zjištěno fetální úmrtí [33].
PATOFYZIOLOGICKÉ MECHANISMY JEDNOTLIVÝCH TMA
Gestační hypertenze/preeklampsie
Při stanovení diagnózy hypertenze po 20. týdnu těhotenství se zatím z neznámých důvodů u jedné skupiny těhotných prezentují „mírné“ změny zahrnující pouze hypertenzi a u druhé se prezentuje klinicky preeklampsie. U mnoha těhotných se vyvine z gestační hypertenze preeklampsie, která je na rozdíl od gestační hypertenze již zařazována mezi TMA. V případě preeklampsie se jako další diagnostické kritérium přidává proteinurie a/nebo projevy orgánové dysfunkce. Ty mohou být velmi různorodé a zahrnují projevy endotelové dysfukce spojené s postižením periferní mikrocirkulace obecně (trombocytopenie a MAHA), mikrocirkulace ledvin, mozku, plic [1]. V obou případech se jedná o stavy, které lze považovat z větší části za reverzibilní neboli dočasné diagnózy.
Podle nejnovějších dat lze u obou typů hypertenzního onemocnění hledat původ v samém úvodu těhotenství – v chybné komunikaci imunitního systému matky s trofoblastem. Důkazy lze dokládat velmi obtížně, protože se tak děje perikoncepčně a mateřské krevní testy ještě nemusí zachytit tak diskrétní změny. Dílčí práce již však na uvedená fakta ukazují. Garcia Salazar se zabývala tím, zda je možné u žen, u kterých se později vyvine preeklampsie, zachytit změny v buněčné imunitě již v úvodu těhotenství a prokázala zjevné prozánětlivé nastavení fenotypu T lymfocytů (převaha počtu Th-1 a Th-17 lymfocytů proti T regulačním lymfocytům) [11]. Podle dostupných údajů je pravděpodobné, že predispozice k vývoji preeklampsie může být dána ještě prekoncepčně.
Za jednu z klíčových buněčných populací zodpovědných za remodelaci spirálních arterií při invazi trofoblastu do deciduy jsou považovány NK buňky (NK – natural killers cells, přirození zabíječi, subpopulace lymfocytů). Pro jejich správnou funkci jsou důležité povrchové KIR receptory (killer imunoglobulin receptor) NK buněk. Jejich exprese je dána správnou balancí exprese inhibičních a aktivačních KIR genů. Moffett ve své práci uvádí, že u žen s preeklampsií je nalézána významně častější exprese inhibičních KIR genů s tím, že uvedené v důsledku vede k nesprávné funkci KIR receptoru, následně chybné remodelaci spirálních arterií a nakonec i k nesprávnému vývoji placenty [23]. Přitom, jak uvádí ve své další práci, jistě zde bude hrát roli i prezentace konkrétních fetálních alel otcovského původu. Konstelace mateřského KIR AA inhibičního haplotypu a paternálního HLA-C2 haplotypu s sebou nese vysoké riziko abnormální placentace, se všemi možnými důsledky, jako jsou například růstová restrikce plodu či preeklampsie [24]. Až velmi pozdním důsledkem chybné „materno-fetální“ komunikace je abnormální remodelace spirálních arterií deciduy a rozvoj endotelové dysfukce, charakterizované mj. výkyvy hladin antiangiogenních (sFlt-1, s-eng) a angiogenních (PlGF, PAPP-A) látek. Tyto parametry jsme schopni identifikovat již od desátého týdne těhotenství a změny jejich hladin se mohou používat k predikci preeklampsie v klinické praxi [3, 9].
Endotelová dysfunkce se týká jak systémového oběhu matky, tak i mikrocirkulace placenty. Vzhledem k patogenezi, která stojí v pozadí obou diagnóz, je nutné monitorovat funkci ledvin, jater, krevní obraz a srážlivost krve.
V průběhu těhotenství přirozeně stoupá clearance kreatininu, proto je velmi důležité sledovat dynamiku jeho hladin. Za horní mez fyziologických hladin je totiž považována hodnota přibližně 80 µmol/l [14]. Hodnocení změn hladin kreatininu v průběhu těhotenství představuje citlivější ukazatel glomerulární filtrace ledvin než „tradičně“ uváděné hodnoty kyseliny močové. Progresivní vzestup hladin kreatininu nad uvedenou mez u gestační hypertenze/preeklampsie může ukazovat na projevy postižení mikrocirkulace ledvin. Hlavním záměrem tohoto článku je podat přehled o problematice, a ne konkrétní klinický algoritmus v případě gestační hypertenze a preeklampsie. Podrobné údaje jsou předmětem Doporučeného postupu ČGPS „Hypertenzní onemocnění v těhotenství“ [39].
Obecně platí, že dobře korigovaná gestační hypertenze s sebou nese možnost ambulantního sledování a pravidelné kontroly laboratorních ukazatelů funkce ledvin, jater, iontogramu, srážlivosti, krevního obrazu, selfmonitoring krevního tlaku a pravidelné hodnocení růstu plodu. Při dobré kompenzaci hypertenze a absenci růstové restrikce plodu preferujeme porod v termínu a plánování porodu podle daných podmínek po dosažení termínu. V případě preeklampsie volíme hospitalizaci, cílem našeho managementu jsou identická opatření při gestační hypertenzi a při absenci ohrožení matky a/nebo plodu se snažíme prodloužit těhotenství. Konkrétní péče závisí na řadě ukazatelů, v případě progrese preeklampsie do obrazu onemocnění „s těžkými rysy“ (TK vyšší než 160/110, známky orgánové dysfunkce apod.), plánujeme porod podle daných podmínek v nejkratší možné době, samotná preeklampsie ale nepředstavuje kontraindikaci vaginálního vedení porodu.
Po porodu ženám doporučíme dispenzární péči praktickým lékařem a po skončení šestinedělí je vhodné verifikovat, zda je přítomna proteinurie/dysfunkce ledvin. Přetrvává-li nadále, ex post nemůžeme takovou diagnózu označit jako preeklampsii, ale jako sekundární hypertenzi nasedající často na jinou diagnózu – především skrytou nefropatii. Jak ukazují recentní data, dá se očekávat, že přes úpravu krevního tlaku po skončení šestinedělí přetrvává vliv endotelové dysfunkce/postižení mikrocirkulace a remodelace myokardu na pozdější zdraví matky a pravděpodobně i dítěte [4, 39].
HELLP syndrom
S přibývajícími poznatky je zřejmé, že se v patogenezi TMA vyskytují mutace a polymorfismy genů kódujících proteiny účastnící se imunitních procesů v orgranismu. Podle dostupných poznatků je zjevné, že i HELLP syndrom je spojen s mutacemi genů kódujících proteiny komplementu a také je již řazen mezi TMA [38]. HELLP syndrom je stanoven jako pracovní diagnóza, ale její potvrzení můžeme učinit, odezní-li projevy a laboratorní příznaky do 48 až 72 hodin po porodu. Diagnostická kritéria HELLP syndromu jsou laboratorní, a přestože jsou všeobecně dobře známa, je vhodné uvádět i širší kontexty informací, mj. právě z pohledu diferenciální diagnostiky. Hemolýza se u HELLP syndromu projevuje mikroangiopatickou hemolytickou anémií, spojenou s přítomností schistocytů v krevním obraze, hyperbilirubinémií a poklesem hladiny haptoglobinu, který vyvazuje hemoglobin z poškozených erytrocytů. Elevace transamináz se týká především AST (hepatocelulární poškození), ale obvyklé je zachytit i elevaci ALT. Trombocytopenie je nejvýznamnějším prognostickým ukazatelem diagnózy a dále ji klasifikuje Mississippi Triple-class HELLP System. Dalšími doprovodnými laboratorními nálezy mohou být elevace CRP a pokles hladiny antitrombinu – AT III (obojí obraz endotelové dysfunkce) [21, 38].
Také klinické projevy jsou velmi nespecifické a zahrnují kromě bolestí v epigastriu a nauzey a/nebo zvracení také „flu-like“ obtíže připomínající virózu, a to včetně subfebrilií. Klinické obtíže mají zpravidla progresivní charakter, nemusí však být přítomny. Incidence HELLP syndromu je asi 2–6 případů na 1000 těhotných. Je důležité vědět, že u třetiny žen se vyvine do 48 hodin po porodu [37]. Právě skupina žen, u nichž se vyvine HELLP syndrom, vyžaduje velmi bedlivé sledování, protože může zahrnovat i pacientky, u nichž se o HELLP syndrom nejedná a může to být obraz jiných trombotických mikroangiopatií. Obecně platí, že by u HELLP syndromu mělo dojít ke klinické a laboratorní regresi nejpozději do 72 hodin po porodu. Pokud se tomu tak neděje, je nutné pomýšlet na jiné TMA – především na získanou formu TTP, HUS/aHUS a akutní těhotenskou steatózu jater (AFLP – acute fatty liver of pregnancy). U těchto diagnóz, které budou popsány níže, jsou laboratorní změny a často i příznaky velmi podobné jako u HELLP syndromu.
Trombotická trombocytopenická purpura
Trombotická trombocytopenická purpura (TTP) je vzácné, klinicky závažné onemocnění ze skupiny trombotických mikroangiopatií s vysokou mortalitou bez včasné adekvátní léčby. Je charakterizována pentádou příznaků (trombocytopenie, mikroangiopatická hemolytická anémie, neurologické příznaky, horečka a renální selhání), která však ve většině případů není kompletně vyjádřena.
Příčinou onemocnění je těžký deficit depolymerázy (metaloproteázy) von Willebrandova faktoru (vWf) ADAMTS13 [16]. Laboratorní stanovení deficitu je součástí diagnostických kritérií. Ve většině případů se jedná o získaný deficit vznikající především v dospělosti a vyvolaný vznikem autoprotilátek proti enzymu. Vzácný vrozený deficit je způsoben mutacemi v genu pro ADAMTS13 (Upshawův-Schülmanův syndrom). Aktivitu metaloproteázy ADAMTS13 je možné stanovovat již i v podmínkách českých laboratoří a za závažnou formu je považována aktivita nižší než 5 %. Pokud se TTP poprvé objeví během těhotenství (získaná forma) nebo se jedná o relaps již stanovené diagnózy, představuje závažný stav spojený s vysokým rizikem ohrožení matky (multiorgánové selhání) a plodu (riziko fetálního úmrtí) [31, 32]. Péče patří především do rukou hematologa. Máme-li před sebou obraz HELLP syndromu, jehož obraz neodeznívá do 48–72 hodin do porodu, měli bychom na TTP pomýšlet a provést laboratorní vyšetření na aktivitu ADAMTS13. Opatření jsou při TTP zatím spíše nespecifická a spočívají v plazmaferéze (PPX – plasma exchange), podání kortikoidů za účelem imunosuprese, popřípadě je nyní k dispozici i biologická léčba cílící na receptor CD20 na B lymfocytech – rituximab [10].
Hemolyticko-uremický syndrom
Termín hemolyticko-uremický syndrom (HUS, STEC-HUS) byl použit poprvé švýcarským hematologem Gasserem a jeho spolupracovníky, kteří popsali klinické příznaky tohoto onemocnění u pěti dětí v roce 1955 [12]. Nemoc je charakterizována triádou příznaků: neimunitní Coombs negativní mikroangiopaticko-hemolytickou anémií, trombocytopenií a akutním renálním selháním. HUS je nejčastější příčinou akutního selhání ledvin v dětském věku, nicméně objevuje se i v období dospělosti [8]. Patogeneze zde ještě není zcela objasněna, ale podle poslední klasifikace je možné spouštěcí příčiny typické formy HUS rozdělit do dvou základních skupin. Nejvýznamnější jsou infekční, virové – H1N1 chřipka, HIV, chřipka typu A, další bakteriální – Streptococcus pneumoniae a enterobakterie produkující shiga-toxin (tzv. shiga toxin-producing Escherichia coli – STEC-HUS). Méně často se v literatuře citují jiné příčiny, konkrétně HUS spojený s transplantací kostní dřeně, solidních orgánů, malignitou, autoimunitou, léky či provázející závažnou hypertenzi a jiné preexistující renální onemocnění. Mezi nejvýznamnější a nejčastější patří HUS provázející infekci shiga-toxin produkujícími kmeny Escherichia coli (STEC, sérotypy O157:H7 a O104:H4). Prvním projevem infekce těmito mikroorganismy je krvavý průjem a relativně často se v jeho průběhu rozvíjí HUS, který je při průkazu shiga-toxinu označen jako STEC-HUS. Patogenetické mechanismy u typické formy HUS nejsou zatím prozkoumány, ale velmi pravděpodobně budou souviset s dysregulací komplementu. Ve vyšetřovacím diferenciálně diagnostickém algoritmu TMA je tak potřeba mj. stanovit přítomnost shiga-toxinu v séru pacienta [26].
Atypická forma hemolyticko-uremického syndromu
Nejvýznamnější trombotickou mikroangiopatií, kterou zvažujeme jako diferenciálně diagnostickou možnost k HELLP syndromu, je atypická forma hemolyticko-uremického syndromu (aHUS). V těhotenství dochází k přirozené aktivaci komplementu, složky vrozené imunity. Aktivační složky komplementu jsou za fyziologických podmínek v přirozené rovnováze s jeho inhibitory. Podle dostupných literárních zdrojů, a především podle autorit v oboru, je patogeneze aHUS chápána jako dysregulace této rovnováhy [27, 28]. Diagnóza aHUS je však stanovena až „per exclusionem“ po vyloučení jiných trombotických mikroangiopatií, především TTP a HUS. Neprokazujeme tedy ani snížení aktivity ADAMTS13, ani není přítomen shiga-toxin v séru a nejsou ani jiné další zjevné souvislosti uvedené v kapitole o HUS [6]. V patogenezi aHUS je nejčastěji popisován deficit faktorů regulující komplement, jako jsou complement factor H (CFH), complement factor H related proteins (CFHR), complement factor I (CFI). Nejčastěji shledáváme deficit regulačního proteinu, který se nazývá membrane cofactor protein (MCP, CD46). Relativně méně často nalézáme abnormality v proteinech, které mohou akcelerovat alternativní dráhu komplementu – complement factor B (CFB), C3 složka komplementu. Dochází k neregulované amplifikaci komplementu, která vede k masivní tvorbě C5 složky komplementu, formaci membrane attack complex (MAC) a k lýze/poškození buněk. Na konci této kaskády je pak „autoagresivní“ poškození endotelu, mikroangiopatická hemolytická anémie, trombocytopenie a orgánová dysfunkce [25].
Přestože se dosud jeví incidence aHUS ve vyspělých zemích zatím velmi nízká (incidence v Evropě 0,2 případů na milion lidí), autority se v literatuře shodují na tom, že vzhledem k přesnější diagnostice se dá očekávat relativní nárůst incidence, a to především u těhotných žen a rodiček. Donedávna totiž nebyly k dispozici současné diagnostické a terapeutické možnosti. Řada těhotných/rodiček byla považována za pacientky s HELLP syndromem, přestože k regresi laboratorních a klinických příznaků do 48–72 hodin nedocházelo. V nemalém počtu případů tak s sebou takové situace nesly i úmrtí pacientek a přinejmenším dlouhodobou morbiditu. Postižení ledvin je často trvalé, od chronické renální insuficience až po potřebu transplantace ledvin [6]. Je tedy možné se pokusit odlišit aHUS od HELLP syndromu na podkladě „robustnosti“ klinických projevů? Na rozdíl od HELLP syndromu (zde především patrné postižení jater) bývají klinické příznaky orgánové dysfunkce více vyjádřeny. Součástí klinického obrazu mohou být příznaky neurologické (zmatenost, encefalopatie, cévní mozková příhoda, křeče aj.), gastrointestinální (nauzea, zvracení, pankreatitida, kolitida), renální projevy (obraz renální insuficience) a kardiovaskulární postižení (hypertenze, trombóza). Jedná se však o soubor nespecifických projevů a konečná diagnóza aHUS se musí učinit až po vyloučení jiných příčin TMA. V literatuře se také více diskutuje o podskupině aHUS pregnancy associated aHUS (P-aHUS) jako o nozologické jednotce, kterou je třeba především kvůli vysokému riziku mortality či morbidity matek od HELLP odlišit [7]. Pokud pomýšlíme na diagnózu aHUS, tj. neodeznívají-li projevy HELLP syndromu do 48–72 hodin po porodu, je kromě výše uvedených vyšetření krve matky na shiga-toxin a ADAMTS13 vhodné odebrat panel regulačních proteinů komplementu, jsou-li v možnostech daného pracoviště (jedná se o stanovení výše uvedených faktorů).
Jsou-li vyšetření provedena a máme-li podezření na aHUS, prvním klíčovým léčebným opatřením je plazmaferéza. Nedochází-li po plazmaferéze k restituci laboratorního a klinického obrazu a/nebo jsou-li prokázány abnormality v proteinech komplementu, je v současné době k dispozici monoklonální protilátka proti C5 složce komplementu, eculizumab. Tato biologická léčba zabraňuje blokádou C5 složky komplementu formaci MAC, a tímto způsobem nastavuje rovnováhu dysregulovaného komplementu. Přestože zatím nemáme k dispozici přesný algoritmus jeho podání pacientům, terapie eculizumabem velmi účinně stabilizuje aHUS a snížuje mortalitu i morbiditu pacientů [28, 34].
Akutní těhotenská steatóza jater
Akutní těhotenská steatóza jater – AFLP (acute fatty liver of pregnancy) je nově řazena mezi trombotické mikroangiopatie. Je to velmi závažná diagnóza charakterizovaná dysfunkcí nebo již selháním jater těhotné/rodičky, které mohou vést ke komplikacím, k ohrožení života matky a plodu, včetně smrti. Pokud na AFLP pomýšlíme v těhotenství, je nutné velmi rychle plánovat ukončení těhotenství a následuje podpůrná péče o matku.
Etiopatogeneze není přesně známa, předpokládá se abnormální metabolismus mastných kyselin na straně plodu. Přibližně 20 % případů má doloženo deficit fetálního „long-chain 3-hydroxyacyl CoA dehydrogenase“ (LCHAD), jednoho z enzymů účastnícího se oxidace mastných kyselin [36]. Vzhledem k tomu, že u většiny pacientů není možné deficit enzymů beta oxidace mastných kyselin prokázat, zatím není zřejmé, jakými mechanismy porucha ovliňuje matku, ale obecně se předpokládá toxické ovlivnění hepatocytů matky intermediárními produkty z fetální cirkulace [15]. Rizikovými faktory pro AFLP je mužské pohlaví, vícečetné těhotenství, nízký body mass index (pod 20), již diagnostikovaná preeklampsie a předchozí AFLP. Incidence se pohybuje přibližně kolem jednoho na 7000–20 000 těhotenství [20, 22].
Klinicky se u pacientek prezentuje jako fulminantní jaterní selhání, se všemi souvislostmi s tím spojenými. Často je zjevný ikterus, únava, v anamnéze je polydipsie, polyurie. V laboratorních nálezech bývá přítomen „neúplný“ obraz HELLP syndromu s trendem k poklesu počtu trombocytů. Jak je uvedeno níže, trombocytopenie není součastí diagnostických kritérií AFLP. Mnohdy se pacientky dostaví pro absenci vnímání pohybů plodu a může být potvrzeno intrauterinní fetální úmrtí. Podobně jako jiné trombotické mikroangiopatie, především aHUS, je diagnóza AFLP stanovena často až per exlusionem, nicméně pro velmi vysoké riziko ohrožení života matky je naléhavě nutné pokusit se k diagnóze dospět co nejdříve. Pro určení pracovní a posléze i definitivní diagnózy je doporučeno využít kritéria Swansea [18]. Splňuje-li pacientka šest a více kritérií níže uvedených, potvrzuje to diagnózu AFLP.
- Kritéria Swansea
- Zvracení
- Bolesti břicha
- Polydipsie/polyurie
- Encefalopatie
- Elevace celkového bilirubinu více než 14 µmol/l
- Glykémie méně než 4 mmol/l
- Elevace kyseliny močové více než 340 µmol/l
- Leukocytóza nad 11 tisíc
- Ascites a/nebo obraz steatózy jater podle UZ vyšetření
- Elevace AST a/nebo ALT
- Elevace amoniaku v séru nad 47 µmol/l
- Renální insuficience – kreatinin více než 150 µmol/l
- Koagulopatie – prodloužení APPT, INR, deficit fibrinogenu
- Mikrovezikulární steatóza jater z biopsie
Management AFLP je založen především na promptním ukončení těhotenství. V diferenciální diagnostice je potřeba vyloučit jiné příčiny TMA, paralelně podle přítomných kritérií Swansea zvažujeme tuto diagnózu. Matka je při jaterním selháním nejvíce ohrožena koagulopatií typu DIC (diseminovaná intravaskulární koagulopatie) – játra nejsou schopna adekvátně syntetizovat koagulační faktory. Jako u jiných TMA je nutná mezioborová spolupráce s tím, že život zachraňujícím opatřením při AFLP je substituce koagulačních faktorů, plazmy a fibrinogenu.
VLASTNÍ POZOROVÁNÍ
Kazuistika 1
Devatenáctiletá primigravida primipara byla přijata do intermediárního centra in gr. hebd. 33+4 pro hrozící předčasný porod. Při přijetí jí byla podána tokolýza betamimetiky a současně kortikoidy k indukci plicní zralosti. Pacientka neměla řádnou prenatální péči, jedenkrát za těhotenství byla ve II. trimestru vyšetřena laboratorně, k dispozici byl výsledek krevního obrazu z 28. týdne, kde byl kromě trombocytopenie (113 tisíc trombocytů) normální krevní obraz. Z anamnézy byl významným údajem abúzus pervitinu a THC, ale jinak byla zcela zdráva, bez jakýchkoliv zdravotních komplikací. Z laboratorních vyšetření při přijetí byl přítomen „obraz HELLP syndromu“ s koagulopatií. V krevním obraze byla anémie, leukocytóza a trombocytopenie – Leu 21 tis., Hgb 118 g/l, Plt 84 tis., v biochemických vyšetřeních kreatinin 187 mmol/l, bilirubin 72 mmol/l, ALT 2,73 µkat/l, AST 2,32 µkat/l, z koagulačních vyšetření APTT 80 s, INR, 1,98, fibrinogen 0,59 g/l, ostatní parametry v mezích normy. Po vysazení tokolýzy proběhl spontánní předčasný porod s epiziotomií a krevní ztrátou 1500 ml. Pro uvedený laboratorní obraz a současně krevní ztrátu při porodu proběhla substituce erymasami (čtyřikrát), plazmou a fibrinogenem. Po porodu byla opakovaně provedena resutura epiziotomie pro tvorbu hematomu. Současně opakovaně metroragie nereagující na konzervativní léčbu uterotoniky. Pro přetrvávající a neustupující „obraz HELLP syndromu“, opakované metroragie, hematomy v ráně a nově i obraz suspektního hemoperitonea podle UZ vyšetření byla přeložena třetí den po porodu do perinatologického centra Gynekologicko-porodnické kliniky 1. LF UK a VFN. Vybrané laboratorní ukazatele při přijetí (ostatní laboratorní vyšetření byla v normě):
- Krevní obraz: Leu 51 tis, Hgb 75 g/l, Plt 68 tis.
- Biochemie: urea 8 mmol/l, kreatinin 187 mmol/l, bilirubin celkový 75 mmol/l, ALT 0,93 µkat/l, AST 1,38 µkat/l, LD 19 µkat/l, CRP 13, haptoglobin méně než 0,06 g/l.
- Koagulační vyšetření: APTT 41, INR 1,2, fibrinogen 2,4 g/l.
Vzhledem k uvedeným laboratorním vyšetřením a celkovému klinickému stavu už nemůžeme třetí den po porodu pacientku vést pod obrazem suspektního HELLP syndromu a vyslovujeme podezření na jinou TMA. Je přítomen obraz hemolytické anémie – MAHA (elevace LD, „neměřitelný“ haptoglobin, hraniční počet schistocytů, Coombsův test dodatečně negativní), trombocytopenie, koagulopatie, známky dysfunkce více orgánů – akutní renální insuficience, hepatocelulární léze, extrémní leukocytóza, navíc ještě projevy hyperhydratace, capillary leak – pravostranný pleurální výpotek a ascites s periferními otoky. Pro zjištěný závažný klinický a laboratorní obraz byla pacientka přeložena na Kliniku anesteziologie, resuscitace a intenzivní medicíny (KARIM). Vytvořen byl mezioborový tým, v němž kromě anesteziologa a porodníka byl i nefrolog a hematolog.
Odebrán ADAMTS13 – za 48 hodin po odběru aktivita enzymu 31 %, tj. normální, vylučujeme tak TTP. V den přijetí již byla provedena první plazmaferéza a zahájeno podávání kortikoidů, shiga-toxin negativní vylučuje STEC-HUS. Odebrány jsou proteiny komplementu, vyslovujeme podezření na atypickou formu HUS. Během hospitalizace na KARIM, čtvrtý den po porodu, byla pacientka pro rozvoj neurologické symptomatologie (tonické křeče) zaintubována a zajištěna sedací, CT mozku vstupně bez patologických změn, s odstupem na kontrolním CT osmý den již počínající edém mozku. Šestý den po porodu při UZ vyšetření dělohy byla zjištěna lochiometra a vaginální cestou (instrumentální revizí děložní dutiny v celkové anestezii) bylo vybaveno 800 ml koagul, substituce krevními deriváty (šestkrát erymasa, šest jednotek mražené plazmy, 2 g fibrinogenu). Postupně byl stav stabilizován, pro oligoanurické renální selhání byla přechodně zahájena dialýza, ukončena byla osmý den po porodu. Od osmého dne po porodu byla denně plazmaferéza. Hypertenze korigována parenterálními antihypertenzivy (dihydralazin a ebrantil). Pravostranná hrudní drenáž, punkce ascitu (5l hemoragický výpotek, kultivačně negativní). Empiricky od osmého dne ATB terapie (meropenem) eskalována pro vzestup parametrů zánětu (+ linezolid a flukonazol, kultivačně v bronchoalveolární laváži kvasinky) do 15. dne. Jedenáctý den po porodu byla extubována, kontrolní CT již s normálním nálezem na mozku. Vyšetřena byla 15. den po porodu porodníkem pro bolest v podbřišku a významný odchod lochií, v děloze přítomna koagula (stacionární obraz), nebyla indikována k revizi dělohy, podána uterotonika, odchod očistků již minimální. Během téhož večera ale progrese bolestí břicha, obleněná peristaltika, rozvoj oběhové nestability, na CT potvrzen objemný hematom v retroperitoneu, provedena byla operační revize z dolní střední laparotomie, revize retroperitonea a vyprázdnění koagul, tamponáda rouškami, krevní ztráty odhanuty na přibližně 1500 ml, pacientka je ponechána v intubaci. Od 15. dne nová kúra ATB, empiricky tygecyklin + amikacin + antimykotikum flukonazol (anamnesticky multirezistentní enterobakter včetně odolnosti vůči karbapenemům). Osmnáctý den po operaci extrakce roušek z retroperitonea, bez pokračujícího krvácení, následně večer extubace. Pacientka byla přeložena na oddělení akutní medicíny Nefrologické kliniky OAM VFN.
Devatenáctý den po operaci byla pacientka výrazně zahleněná, nedostatečná svalová síla k adekvátní expektoraci a toaletě dýchacích cest, febrilní, omezeně spolupracující, proto byla při hyposaturaci kyslíkem od 20. dne opět intubována a zahájena byla umělá plicní ventilace. Kontrola pleurálního výpotku vlevo – 1,5cm lem vysoko při vysokém postavení bránice. Provedena bronchoskopická toaleta dýchacích cest, RTG obraz prakticky normalizován, vzdušný plicní parenchym, odeznívající výpotek v pleurální dutině, dosaženo negativní tekutinové bilance. Po snížení sedace 21. den po operaci autoextubace, ventilačně stabilní. K dispozici již výsledek proteinů komplementu – deficit regulačního proteinu komplementu MCP (CD46, viz výše). Pro dg. aHUS (ostaní příčiny TMA vyloučeny) byly 22. den ukončeny denní plazmaferézy a podána první dávka eculizumabu. Od 26. dne po porodu normalizace počtu trombocytů až přechod do výrazné trombocytémie, doplněn ruční diferenciál krevního obrazu a průtoková cytometrie (Fluorescent Activated Cell Sorting – FACS) periferní krve k vyloučení hematologické malignity, zajištěna antiagregační léčbou 100 mg kyseliny acetylsalicylové. Odstranění permanentního močového katétru úspěšné, bez retence. Třicátého dne po porodu překlad na standardní oddělení.
Klinický stav se nadále lepší, pacientka je schopna samostatné chůze, normální per os příjem stravy i tekutin. Od 31. dne zaznamenány intermitentní subfebrilie. CRP opakovaně negativní. Bez přesvědčivých kontrolních kulivačních nálezů. RTG srdce a plic bez patologického nálezu, 34. den kontrolní gynekologické vyšetření s normálním klidným UZ i klinickým nálezem v šestinedělí. Další systémová antibiotická léčba již pacientce nebyla podána. Vzhledem k příznivému laboratornímu a klinickému nálezu bylo rozhodnuto o nepodání další dávky eculizumabu, jelikož původní stimul (porod a pravděpodobně i infekce) již odezněl. Trombocytóza trvá, pravděpodobně restituční obraz původní dysfunkce endotelu. Klinický stav je z hlediska systémového onemocnění t.č. relativně stabilizovaný, postupně je pacientka afebrilní. Po domluvě je přeložena na gynekologicko-porodnické oddělení intermediárního centra.
Při překladu je kardiopulmonálně kompenzovaná, eupnoická, eufrekvenční, tlakový průměr kolem 120/80 mm Hg, poslechový nález na plicích s oslabením vpravo bazálně – dáno vyšším stavem bránice, bez výpotků, břicho nebolestivé, jizva hráze i dolní střední laparotomie zhojena per primam, bez otoků, diuréza hojná, schopna samostatné chůze, reziduálně mírný klidový tremor horních končetin. Z intermediárního centra byla propuštěna za několik dní v dobrém stavu nevyžadujícím hospitalizaci, plně soběstačná. Předána byla do dispenzární péče ambulance Nefrologické kliniky VFN. S odstupem jednoho roku po porodu je pacientka klinicky nadále bez obtíží a laboratorně zcela bez odchylek.
Kazuistika 2
Třicetiletá primigravida primipara byla přijata na porodní sál Gynekologicko-porodnické kliniky 1. LF UK a VFN pro absenci vnímání pohybů plodu v 33. týdnu těhotenství. Klinicky zjevně ikterická, polydipsie/polyurie (sytě žlutá moč), bolesti v podžebří vpravo, vše subjektivně vnímá i objektivně pozoruje partner přibližně týden. V anamnéze astma alergického původu bez léčby. Podle UZ vyšetření nebyly zastiženy ozvy plodu, potvrzen fetus mortuus, eutrofický plod odhadem odpovídající 50% percentilu růstu. Pacientce a partnerovi vysvětlujeme stav a po laboratorních vyšetřeních je v plánu vyvolání porodu mrtvého plodu. V laboratorních vyšetřeních patrné vstupní hodnoty krevního obrazu KO: Leu 33 tisíc, Hb 145 g/l, trombocyty 333 tisíc, dodatečně schistocyty negativní. V biochemických ukazatelích urea 4 mmol/l, kreatinin 100 mmol/l, kyselina močová 430 µmol/l, celk. bil. 122 mmol/l, ALT 7,44 µkat/l, AST 6,62 µkat/l, haptoglobin 0,66 g/l = norma. Koagulační vyšetření – INR 1,8, APTT 60, trombinový čas 26, fibrinogen 0,77 g/l, AT III 11, D-dimery 1613.
Vzhledem k laboratorním vyšetřením a klinickému stavu vyslovujeme podezření na TMA, susp. AFLP a rozhodujeme se přistoupit neprodlenně k ukončení těhotenství císařským řezem z vitální indikace matky. Doplňujeme odběry na ADAMTS13, proteiny komplementu, shiga-toxin. Při vědomí, že je AFLP spojena s koagulopatií na podkladě snížené syntézy koagulačních faktorů játry, po dohodě s anesteziology podáváme celkem šest jednotek plazmy a 4 gramy fibrinogenu. Po nekomplikovaně probíhajícím císařském řezu byl porozen mrtvý hoch váhy 1890 gramů a stav pacientky byl stabilizován na JIP gynekologicko-porodnické kliniky, opakovaně korigována koagulace – podání plazmy, antitrombinu, fibrinogenu. Probíhá korekce vnitřního prostředí (včetně albuminu), pacientce jsou ordinována hepatoprotektiva a nízkomolekulární heparin. Šestého pooperačního dne je stav pacientky stabilizován a je předána do péče hepatologa, kde před dimisí, sedmého dne, uzavíráme po konzultaci s ním jako suspektní akutní těhotenskou steatózu jater (AFLP). Pitevní průkaz placenty a plodových obalů bez významných odchylek, plod jeví projevy úmrtí v důsledku asfyxie.
Pacientka byla propuštěna domů v dobrém stavu s mírnou elevací transamináz, ostatní laboratorní vyšetření v normě. Hepatolog doporučuje jaterní biopsii, která byla provedena v ambulantním režimu 10. den po porodu. Výsledek biopsie potvrzuje podezření na AFLP – mírná odeznívající steatóza jaterní tkáně. Při komplexním hodnocení případu verifikujeme přítomnost osmi kritérií Swansea (bolesti břicha, polyurie/polydipsie, elevace bilirubinu, kyseliny močové, transamináz, leukocytózu, přítomnost koagulopatie a steatózu jater), diagnóza tedy byla po dohodě s hepatology jako AFLP definitivně uzavřena. Z vyšetření proteinů komplementu byl potvrzen deficit MCP (CD46), poukazující na možnost vývoje aHUS v budoucím životě. V dalším průběhu je žena pravidelně sledována hepatology a nefrology, s kompletní úpravou laboratorních ukazatelů.
DISKUSE
Pro mnoho porodníků představuje problematika trombotických mikroangiopatií (TMA) relativně neznámou oblast. Předložili jsme dosavadní poznatky o této problematice a na dvou kazuistikách ukázali praktický přístup, který bychom měli k takovým pacientkám uplatňovat. Obecně je potřeba znovu zdůrazňovat, že přestože nejčastější příčinou hemolýzy, elevace transamináz, trombocytopenie a změny v koagulaci je HELLP syndrom (který se mezi TMA také řadí), je nutné každou takovou situaci pečlivě přehodnocovat, a zejména neodezní-li výše uvedený obraz spontánně do 48–72 hodin po porodu, uvažovat o jiných TMA. V České republice zatím není žádný ucelený algoritmus, který bychom mohli využít, autoři tohoto článku proto navrhují užívat algoritmus z obrázku 1.
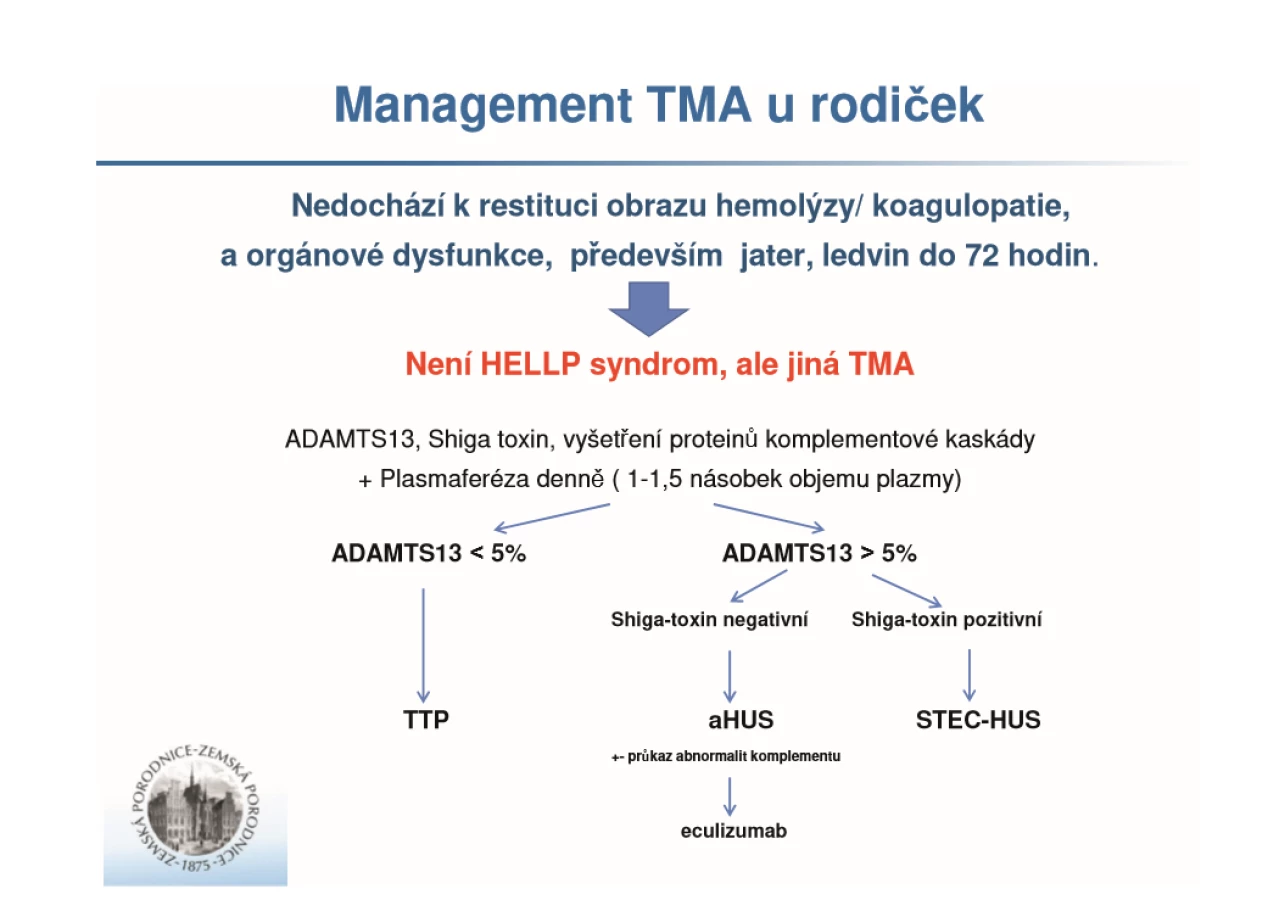
Naše navrhované schéma odráží data prezentovaná v literatuře, kdy uplatňujeme postup per exclusionem [5, 27, 40]. Cílem rychlé diferenciální diagnostiky je především snaha o co nejkratší časovou prodlevu, která by s sebou mohla nést vážné ohrožení života pacientek a/nebo jejich dlouhodobou morbiditu. Paralelně s diagnostickými opatřeními je třeba postupovat vždy ve spolupráci s kolegy, jichž se daná problematika nejvíce týká, tj. anesteziology, nefrology, hematology, hepatology [2]. Konečným opatřením při průkazu aHUS je podání biologické léčby eculizumabu. Pokud není na daném pracovišti k dispozici, je vždy nepodkročitelným minimem plazmaferéza po 5–7 dní. Definitivní zajištění pacientů s jinými TMA s sebou nesou příslušnou symptomatickou léčbu [19]. U AFLP je pracovní diagnóza stanovena relativně velmi rychle, na podkladě základních laboratorních a klinických projevů, a proto není součástí uvedeného schématu. Zde je potřeba co nejrychleji rozhodnout o časném ukončení těhotenství, byť za cenu císařského řezu, a navíc substituovat koagulační faktory, které nejsou poškozenými játry adekvátně syntetizovány.
V první kazuistice předkládáme průběh onemocnění u pacientky, kdy jsme poprvé uplatnili multidisciplinární přístup a uvádíme první zkušenosti s diferenciálně diagnostickým a terapeutickým algoritmem tak, jak jsme jej dosud poznali jen z literárních zdrojů. Kolektiv lékařů pečující o pacientku se při nezkušenosti s péčí o takovou problematiku shodl především na zahájení plazmaferézy; přes toto opatření nedocházelo k restituci klinického a laboratorního obrazu, což vedlo k rozhodnutí o podání eculizumabu. Optimální iniciální a udržovací dávky jsou předmětem diskusí, naše pracoviště nyní pracuje na ucelém návrhu, jak v těchto situacích postupovat. Dostupné literární zdroje se shodují na tom, že je eculizumab vyhrazen pro situace, kde nedochází k odezvě na podání plazmaferézy [29]. Po podání eculizumabu došlu u pacientky k rapidnímu zlepšení a nyní je klinicky zcela bez významných problémů.
Soubor, který prezentuje Alexandra Bruel, však dokládá, jak závažný vliv má diagnóza aHUS na dlouhodobou morbiditu. Její soubor zahrnoval pacientky s aHUS, který se objevil hlavně během prvního těhotenství (58 %) a po porodu (76 %). Při diagnóze vyžadovalo dialýzu 56 (71 %) pacientů; 56 (78 %) pacientů podstoupilo plazmaferézu, 21 (41 %) dostalo infuze plazmy a čtyři (5 %) dostávali eculizumab. Během sledování (průměrné trvání 7,2 roku) dosáhlo 41 (53 %) pacientů ESRD (end-stage renal disease – konečné stadium renálního selhání), 15 (19 %) mělo CKD (chronic kidney disease – chronické renální selhání) a 18 (28 %) pacientů mělo relaps aHUS v dalším průběhu života. Celkem 24 pacientů (27 %) podstoupilo transplantaci ledvin a recidiva aHUS se u nich vyskytla u 13 (54 %). Mutace v komplementových genech byly detekovány u 49 (56 %) pacientů, hlavně u CFH (30 %) a CFI genů (9 %) [6].
V druhé kazuistice prezentujeme případ pacientky, která přichází pod poněkud jiným klinickým obrazem. Uvedená kazuistika dokládá, jak je velmi důležitá anamnéza. Objektivní i subjektivní příznaky jsou totiž důležitými součástmi kritérií AFLP a jejich přítomnost by nás měla v kontextu s laboratorními ukazateli navést na možnou přítomnost této velmi závažné diagnózy. Byť se jedná o vzácnou problematiku, i zde je jakákoliv významná časová prodleva v managementu spojena s narůstem rizika ohrožením matky a plodu. AFLP je spojena s vysokou mortalitou matek nejčastěji na podkladě hemoragie a multiorgánového selhání spojených s deficiencí koagulačních faktorů. Časná intervence, tj. podání vysokých dávek plazmy i fibrinogenu, vedla k poměrně rychlé stabilizaci pacientky.
ZÁVĚR
Trombotické mikroangiopatie představují velmi závažnou heterogenní skupinu syndromů, se kterými se setkáváme nejčastěji v peripartálním období. Spouštěcích příčin může být mnoho, ale bez výjimky je hlavním patogenetickým projevem poškození endotelu ohrožující matku orgánovou dysfunkcí, bezprostřední ohrožení života matky je pak dáno především „krvácivými“ komplikacemi. Definitivní diagnóza je často stanovena mylně, jako obraz suspektního HELLP syndromu. Relativně často se čeká na ústup symptomů a laboratorních změn neadekvátně dlouho, což může vést při časové prodlevě k vystavení pacientky vysokému riziku mortality a dlouhodobé morbidity. V současné době zatím není k dispozici sjednocený algoritmus pro diferenciální diagnostiku a management TMA, ale většina autorit se ve světové literatuře shoduje na postupu, který kolektiv autorů předkládá v tomto článku.
MUDr. Michal Koucký, Ph.D.
Gynekologicko-porodnická klinika
1. LF UK a VFN
Apolinářská 18
128 00 Praha 2
e-mail: michal.koucky@vfn.cz
Zdroje
1. ACOG Practice Bulletin No. 202: Gestational hypertension and preeclampsia. Obstet Gynecol, 2019, 133(1):e1.
2. Afshar-Kharghan, V. Atypical hemolytic uremic syndrome. Hematology Am Soc Hematol Educ Program, 2016, 1, p. 217–225.
3. Ahmed, A. Evidence-based revised view of the pathophysiology of preeclampsia. Adv Exp Med Biol, 2017, 956, p. 355–374.
4. Basit, S., Wohlfahrt, J., Boyd, HA. Pre-eclampsia and risk of dementia later in life: nationwide cohort study. BMJ, 2018, 363, k4109.
5. Berger, BE. Atypical hemolytic uremic syndrome: a syndrome in need of clarity. Clin Kidney J, 2018, 12(3), p. 338–347.
6. Bruel, A., Kavanagh, D., Noris, M, et al. Hemolytic uremic syndrome in pregnancy and postpartum. Clin J Am Soc Nephrol, 2017, 12(8), p. 1237–1247.
7. Burwick, RM., Moyle, KA., Gupta, M. Pregnancy-associated atypical hemolytic uremic syndrome: some answers. Am J Obstet Gynecol, 2019, 220(1), Suppl., p. S397–S398.
8. Fakhouri, F., Zuber, J., Frémeaux-Bacchi, V., Loirat, C. Haemolytic uraemic syndrome. Lancet, 2017, 390(10095), p. 681–696.
9. Fraser, R. Decidual natural killer cells regulate vessel stability: implications for impaired spiral artery remodelling. J Reprod Immunol, 2015, 110, p. 54–60.
10. Fyfe-Brown, A., Clarke, G., Nerenberg, K., et al. Management of pregnancy-associated thrombotic thrombocytopenia purpura. AJP Rep, 2013, 3(1), p. 45.
11. Garcia Salazar, MD., Mobley, Y., Henson, J., et al. Early pregnancy immune biomarkers in peripheral blood may predict preeclampsia, J Reprod Immunol, 2018, 125, p. 25–31.
12. Gasser, C., Gautier, E., Steck, A., et al. Hämolytisch urämische Syndrom: Bilaterale Nierenrindennekrosen bei akuten erworbenen hämolytischen Anämien. Schweiz Med Wchschr, 1955, 85, p. 905–909.
13. Haram, K., Svendsen, E., Abildgaard, U. The HELLP syndrome: Clinical issues and management. A review. BMC Pregnancy Childbirth, 2009, 9, p. 8.
14. Hayslett, JP. Interaction of renal disease and pregnancy. Kidney Int, 1984, 25(3), p. 579.
15. Ibdah, JA., Bennett, MJ., Rinaldo, P., et alk. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med, 1999, 340(22), p. 1723.
16. Joly, BS., Coppo, P., Veyradier, A. Thrombotic thrombocytopenic purpura. Blood, 2017, 129(21), p. 2836–2846.
17. Kappler, S., Ronan-Bentle, S., Graham, A. Thrombotic microangiopathies (TTP, HUS, HELLP). Hematol Oncol Clin North Am, 2017, 31(6), p. 1081–1103.
18. Knight, M., Nelson-Piercy, C., Kurinczuk, JJ., et al. UK Obstetric Surveillance System. A prospective national study of acute fatty liver of pregnancy in the UK. Gut, 2008, 57(7), p. 951.
19. Legendre, CM., Licht, C., Muus, P., et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med, 2013, 368(23), p. 2169–2181.
20. Liu, J., Ghaziani, TT., Wolf, JL. Acute fatty liver disease of pregnancy: updates in pathogenesis, diagnosis, and management. Am J Gastroenterol, 2017, 112(6), p. 838–846.
21. Martin, JN Jr., Rinehart, BK., May, WL., et al. The spectrum of severe preeclampsia: comparative analysis by HELLP (hemolysis, elevated liver enzyme levels, and low platelet count) syndrome classification. Am J Obstet Gynecol, 1990, 180, p. 1373–1384.
22. Minakami, H., Morikawa, M., Yamada, T., et al. Differentiation of acute fatty liver of pregnancy from syndrome of hemolysis, elevated liver enzymes and low platelet counts. J Obstet Gynaecol Res, 2014, 40(3), p. 641–649.
23. Moffett, A. NK cell allorecognition. Nat Rev Immunol, 2017, 17(8), p. 466.
24. Moffett, A., Chazara, O., Colucci, F., Johnson, MH. Variation of maternal KIR and fetal HLA-C genes in reproductive failure: too early for clinical intervention. Reprod Biomed Online, 2016, 33(6), p. 763–769.
25. Noris, M., Caprioli, J., Bresin, E., et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol, 2010, 5(10), p. 1844–1859.
26. Noris, M., Mescia, F., Remuzzi, G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol, 2012, 8(11), p. 622–633.
27. Noris, M., Remuzzi, G. Atypical hemolytic-uremic syndrome. N Engl J Med, 2009, 361, p. 1676–1687.
28. Nürnberger, J., Philipp, T., Witzke, O., et al. Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med, 2009, 360(5), p. 542–544.
29. Raina, R., Grewal, MK., Radhakrishnan, Y., et al. Optimal management of atypical hemolytic uremic disease: challenges and solutions. Int J Nephrol Renovasc Dis, 2019, 12, p. 183–204.
30. Rosove, MH. Thrombotic microangiopathies. Semin Arthritis Rheum, 2014, 43(6), p. 797–805.
31. Scully, M., Hunt, BJ., Benjamin, S., et al. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol, 2012, 158, p. 323–335.
32. Scully, M. Thrombotic thrombocytopenic purpura and atypical hemolytic uremic syndrome microangiopathy in pregnancy. Semin Thromb Hemost, 2016, 42(7), p. 774–779.
33. Stella, CL., Dacus, J., Guzman, E., et al. The diagnostic dilemma of thrombotic thrombocytopenic purpura/hemolytic uremic syndrome in the obstetric triage and emergency department: lessons from 4 tertiary hospitals. Am J Obstet Gynecol, 2009, 200(4), p. 381.e1–6.
34. Stefanovic, V. The extended use of eculizumab in pregnancy and complement activation? Associated diseases affecting maternal, fetal and neonatal kidneys – the future is now? J Clin Med, 2019, 8(3), pii: E407.
35. Theilen, LH., Meeks, H., Fraser, A., et al. Long-term mortality risk and life expectancy following recurrent hypertensive disease of pregnancy. Am J Obstet Gynecol, 2018, pii: S0002-9378(18)30279-5.
36. Thomas, MR., Robinson, S., Scully, MA. How we manage thrombotic microangiopathies in pregnancy. Br J Haematol, 2016, 173(6), p. 821–830.
37. Trávniková, M., Gumulec, J., Kořístek, Z., et al. HELLP syndrome requiring therapeutic plasma exchange due to progression to multiple organ dysfunction syndrome with predominant encephalopathy, respiratory and renal insufficiency. Čes Gynek, 2017, 82(3), p. 202–205.
38. Vaught, AJ., Braunstein, EM., Jasem, J., et al. Germline mutations in the alternative pathway of complement predispose to HELLP syndrome. JCI Insight, 2018, 3(6). pii: 99128.
39. Vlk, R., Procházka, M. Hypertenzní onemocnění v těhotenství. Čes Gynek, 2018, 83, p. 145–154.
40. Zipfel, PF. Thrombotic microangiopathies: new insights and new challenges. Curr Opinion Nephrol Hypertens, 2010, 4, p. 372–378.
Štítky
Dětská gynekologie Gynekologie a porodnictví Reprodukční medicínaČlánek vyšel v časopise
Česká gynekologie

2020 Číslo 1
Nejčtenější v tomto čísle
- Trombotické mikroangiopatie a těhotenství
- Lactobacillus crispatus dominantní vaginální mikrobiota v těhotenství
- Neinvazivní prenatální testy: jejich přínos a limity
- Je možná prevence ovariálního karcinomu?