
Oxytocin a další peptidová uterotonika – jejich pražské osudy
Oxytocin and Further Uterotonic Peptide Agents: Their Early Research in Prague
Amino acid sequence of oxytocin, identified already in 1906 as the uterotonic component of neurohypophyseal extracts, was established in 1953 by Vincent du Vigneaud in New York and Hans Tuppy in Vienna. Its structure was verified by the total synthesis one year after in the du Vigneaud laboratory. In the following years, simplified synthetic strategies elaborated in a number of laboratories worldwide enabled structural modifications of individual sites in the peptide chain, aiming at a detailed elucidation of their influence upon pharmacologic features of oxytocin. Frequently, these peptide analogues opened the way to new, clinically useful drugs. The research on vasopressin, the other main peptide hormone of posterior pituitary, underwent a similar development. Among the first who elaborated a more flexible alternative to du Vigneaud protocol was the peptide group at the Institute of Organic Chemistry and Biochemistry, Czechoslovak Academy of Sciences (ÚOCHB) in Prague, chaired by Josef Rudinger. Its research activities were broadly supported – sometimes even enabled – by František Šorm, director of the Institute. This opened a way to an easier synthesis of oxytocin analogues. Design strategy in Prague was focused on oxytocin analogues with an enhanced metabolic stability (prolongation of half-life in vivo), and on analogues acting as inhibitors to its uterotonic/galactobolic response. In the former case, design strategy has originated from studies of enzymatic stability of oxytocin, accomplished in the biochemical laboratories at the ÚOCHB, or reported in earlier communications. Doseresponse and time-response behaviour of analogues in which potential sites of enzymatic attacks were replaced by resistant sequences, and modified peptides investigated in a number of in situ and/or ex vivo pharmacological experiments. Of particular interest were analogues in which one or both sulphur atoms in the –S-S– bridge were replaced by the methylene group (–CH2–), the so-called carba-analogues. Individual analogues of this series possessed, in various degrees, biological activities of oxytocin but not a prolongation of their responses in pharmacological models or in their physiological clearance. Thus, the carba analogues document, firstly, that the integrity of the disulfide bridge in not a necessary condition of oxytocin (or vasopressin) activity, and secondly, that the –S-S– bridge is not the rate determining site of neurohypophyseal hormone inactivation in vivo. In an attempt to prolong the action of oxytocin, its N-a-group was acylated by an additional amino acid or a short peptide, in expectation that such analogues would act as prohormones: splitting of additional substituent by tissue aminopeptidases would in vivo produce “free” oxytocin (therapeutically, the analogues would act as oxytocin depots). A number of in vivo experiments verified this “hormonogen” model and brought forth some clinically interesting substances; some of them are in use until now. In the latter case, the search for structural modifications potentially leading to antagonism indeed brought some new antagonists but, in particular, contributed to the notion of continuous change from “full” agonism via partial agonism to antagonism, according to the tissue conditions. Such a change could have been achieved for uterotonic response of several analogues by changing calcium and magnesium concentrations in the tissue medium. Ideas originated by Rudinger’s group brought about several clinically useful peptides like Carbetocin, Atosiban, Glypressin, Terlipressin. Very successful was the Prague vasopressin analogue dDAVP (Desmopressin) licensed to the Swedish pharmaceutical company Ferring Läkemedel AB. Josef Rudinger left Czechoslovakia in 1968 and became a professor of molecular biology at the Swiss Federal Institute of Technology (ETH). He passed away, 51 years old, in 1975.
Keywords:
Oxytocin – Vasopressin – neurohypophysis – endocrinology-neurohypophysial hormones – I. P. Pavlov – secretin – carbetocin – dDAVP – deamino-D-arginine vasopressin – desmopressin – atosiban – František Šorm – Josef Rudinger
Autoři:
V. Pliska 1; A. Pařízek 2; M. Flegel†
Působiště autorů:
Collegium Helveticum, Eidgenössische Technische Hochschule (ETH) Zürich & Universität Zürich, Schmelzbergstrasse 25, CH-8092 Zürich
1; Gynekologicko-porodnická klinika 1. lékařské fakulty UK a VFN v Praze, Perinatologické centrum, Apolinářská 18, 128 51 Praha 2
2
Vyšlo v časopise:
Ceska Gynekol 2023; 88(1): 56-66
Kategorie:
Referát
Chem. Listy 2019; 113: 675–685
Souhrn
Amino acid sequence of oxytocin, identified already in 1906 as the uterotonic component of neurohypophyseal extracts, was established in 1953 by Vincent du Vigneaud in New York and Hans Tuppy in Vienna. Its structure was verified by the total synthesis one year after in the du Vigneaud laboratory. In the following years, simplified synthetic strategies elaborated in a number of laboratories worldwide enabled structural modifications of individual sites in the peptide chain, aiming at a detailed elucidation of their influence upon pharmacologic features of oxytocin. Frequently, these peptide analogues opened the way to new, clinically useful drugs. The research on vasopressin, the other main peptide hormone of posterior pituitary, underwent a similar development. Among the first who elaborated a more flexible alternative to du Vigneaud protocol was the peptide group at the Institute of Organic Chemistry and Biochemistry, Czechoslovak Academy of Sciences (ÚOCHB) in Prague, chaired by Josef Rudinger. Its research activities were broadly supported – sometimes even enabled – by František Šorm, director of the Institute. This opened a way to an easier synthesis of oxytocin analogues. Design strategy in Prague was focused on oxytocin analogues with an enhanced metabolic stability (prolongation of half-life in vivo), and on analogues acting as inhibitors to its uterotonic/galactobolic response. In the former case, design strategy has originated from studies of enzymatic stability of oxytocin, accomplished in the biochemical laboratories at the ÚOCHB, or reported in earlier communications. Doseresponse and time-response behaviour of analogues in which potential sites of enzymatic attacks were replaced by resistant sequences, and modified peptides investigated in a number of in situ and/or ex vivo pharmacological experiments. Of particular interest were analogues in which one or both sulphur atoms in the –S-S– bridge were replaced by the methylene group (–CH2–), the so-called carba-analogues. Individual analogues of this series possessed, in various degrees, biological activities of oxytocin but not a prolongation of their responses in pharmacological models or in their physiological clearance. Thus, the carba analogues document, firstly, that the integrity of the disulfide bridge in not a necessary condition of oxytocin (or vasopressin) activity, and secondly, that the –S-S– bridge is not the rate determining site of neurohypophyseal hormone inactivation in vivo. In an attempt to prolong the action of oxytocin, its N-a-group was acylated by an additional amino acid or a short peptide, in expectation that such analogues would act as prohormones: splitting of additional substituent by tissue aminopeptidases would in vivo produce “free” oxytocin (therapeutically, the analogues would act as oxytocin depots). A number of in vivo experiments verified this “hormonogen” model and brought forth some clinically interesting substances; some of them are in use until now. In the latter case, the search for structural modifications potentially leading to antagonism indeed brought some new antagonists but, in particular, contributed to the notion of continuous change from “full” agonism via partial agonism to antagonism, according to the tissue conditions. Such a change could have been achieved for uterotonic response of several analogues by changing calcium and magnesium concentrations in the tissue medium. Ideas originated by Rudinger’s group brought about several clinically useful peptides like Carbetocin, Atosiban, Glypressin, Terlipressin. Very successful was the Prague vasopressin analogue dDAVP (Desmopressin) licensed to the Swedish pharmaceutical company Ferring Läkemedel AB. Josef Rudinger left Czechoslovakia in 1968 and became a professor of molecular biology at the Swiss Federal Institute of Technology (ETH). He passed away, 51 years old, in 1975.
Klíčová slova:
Oxytocin – Vasopressin – neurohypofýza – endokrinologie-neurohypofyzární hormony – I. P. Pavlov – sekretin – carbetocin – dDAVP – deamino-D-arginin vasopressin – desmopressin – atosiban – František Šorm – Josef Rudinger
Historická poznámka: Hormony neurohypofýzy a počátky endokrinologie
Endokrinologie jako autonomní obor biomediciny byla ustanovena na zlomu 19. a 20. století, převážně zásluhou britských fyziologů. Pojem hormon poprvé použil v přednášce (Croonian Lecture) pro Royal College of Physicians of London v červnu 1905 fyziolog Ernest Henry Starling (1866–1927) [1]. Hormonem označil látku produkovanou sekretorickou tkání, a která je transportovaná krevním oběhem do cílové tkáně, kde vyvolává funkční změnya.
Starling tuto definici doložil pokusy se sekrecí pankreatu, které spolu s dalším významným londýnským fyziologem, Williamem Maddock Baylissem (1860–1924), publikovali v roce 1902 [2]. Prokázali, že sekrece pankreatu, kterou vyvolali stimulací tenkého střeva (duodena) kyselinou chlorovodíkovou, trvá i po úplné denervaci střeva. Dále zjistili, že extrakt takto stimulované střevní tkáně vyvolá i po jeho intravenózním podání u pankreatu stejnou sekretorickou reakci jako stimulace kyselinou či postprandiální stimulace duodena potravou. Tento první přesný popis humorální regulace, jako alternativy nervové regulace, měl mnohem širší význam v dalších výzkumech fyziologických procesů. Hypotetickou, částečně ovšem už tehdy předčištěnou stimulující látku, označili jako sekretin. Sekretin se proto v literatuře označuje jako první prokázaný hormonb.
Ne ovšem zcela právem. Biologické účinky látek, později podle Starlingovy a Baylissovy definice identifikované jako hormony, byly totiž známy už dříve. V roce 1888 popsali Oskar Minkowski (1858–1931) a Josef (též Joseph) Freiherr von Mering (1849–1908) [3] glykosurii u psů po odstranění pankreatu a otevřeli tím i cestu k pozdější identifikaci a izolaci inzulinu. V souvislosti s tehdejším zájmem o tzv. organoterapiic studovali o několik let později, v roce 1895, londýnský praktický lékař George Oliver (1841–1915) a fyziolog Edward Albert Schäfer (též Sir Edward Albert Sharpley-Schafer, 1850–1935), pozdější profesor fyziologie na univerzitěv Edinburghu, fyziologické účinky extraktů nadledvinek, štítné žlázy, sleziny a hypofýzy [4,5]. Zajímaly je jejich centrální účinky, u extraktů hypofýzy a nadledvinek především na zvýšení arteriálního tlaku v důsledku vasokonstrikce. William Harry Howell (1860–1945) [6], profesor fyziologie na John-Hopkins University, E. A. Schäfer a (Thomas) Swale Vincentd (1868–1933) [7] pak lokalizovali účinné složky extraktů zadního laloku hypofýzy (1898–1899). Lze tedy říci, že Oliver, Schäfer, Howell a Vincent zavedli jako první do endokrinologického výzkumu biochemicko-farmakologické postupy. Funkce jednotlivých orgánů či tkání byly doposud studovány jejich chirurgickým odnětím (-ektomie). Hormony produkované neurohypofýzou oxytocin a vasopresin (později označovaný jako antidiuretický hormon – AHD) se tak významně zařadily do dějin endokrinologie.
Uterokinetický účinek extraktů hypofýzy pak poprvé popsal v roce 1906 londýnský biochemik Sir Henry Hallett Dale (1875–1968) [8], laktační efekt Isaac Ott (1847–1916) a John C. Scott [9] z Medico-Chirurgical College ve Philadelphii. Peptidový charakter uterokinetické složky extraktů z neurohypofýzy se snažil prokázat Harold Ward Dudley (1887–1935) jejich inaktivací proteolytickými enzymy [10]. Avšak z pohledu dnešní enzymologie šlo spíše o závěry pokusů typu „right-for-the-wrong-reason“. Čisté enzymové preparáty ještě nebyly ve dvacátých létech 20. století většinou dostupné, proto Dudley referuje o rychlé inaktivaci extraktu z neurohypofýzy trypsinem na krysí děloze. Ve svých pokusech používal ale Pankreatin firmy Merck, tedy extrakt pankreatických enzymů z různých živočišných druhů. Pozdější měření potvrdily, že oxytocin není enzymově čistým trypsinem štěpitelný. Dudleyovy výsledky by bylo možno vysvětlit enzymologicky netypickým štěpením oxytocinu mezi oběma koncovými aminokyselinami (leucinem v poloze 8 a glycinamidem v poloze 9) chymotrypsinem [11], neboť chymotrypsin byl totiž téměř jistě v použitém preparátu obsažen.
Tuto průkopnickou epochu pak uzavírá pečlivá práce výzkumníků z laboratoří firmy Parke, Davis and Co. v Detroitu (Michigan). Oliver Kamm se spolupracovníky [12] zde rozdělili v roce 1928 složitým pracovním postupem – opakovanou precipitací, vysolováním a extrakcí do organických rozpouštědel – neurohypofyzární extrakt na dvě frakce, jedna vykazovala převážně uterokinetickou a druhá více vazokonstrikční aktivitu. Dále odhadli (z rychlosti difuze) molekulovou hmotnost uretokinetické substance na 600 Da (později přesně potvrzená molekulová hmotnost oxytocinu činí 1 007 Da) a naznačili, že vazokonstrikční komponenta vykazuje též antidiuretickou aktivitu. Patrně ještě vyšší čistoty obou frakcí dosáhli v polovině třicátých let R. M. Stehle a A. L. Fraser [13]. Protiproudním vytřepáváním v systému voda/2-butanol (techniku zavedl Lyman C. Craig v roce 1944, cit. [14]) získal pak Arthur H. Livermore a Vincent du Vigneaud [15] čistou substanci s vysokou uterokinetickou aktivitou. Tento preparát použil pak na počátku 50. let du Vigneaud (1901–1978) a jeho spolupracovníci tzv. Edmanovým odbouráváním [16] k určení struktury a jako standard při syntéze oxytocinu [17,18].
Du Vigneaud a jeho početný tým spolupracovníků z Cornell University Medical College (Ithaka, NY) má pak jednoznačnou zásluhu na první chemické syntéze vasopresinu a oxytocinu. Tato du Vigneaudova práce byla záhy, v roce 1955, oceněna Nobelovou cenou za chemii. Laudatio Nobelovy komise plně vystihuje jeho zásluhy – a zároveň ovšem i zásluhy jeho spolupracovníků: „… for his work on biochemically important sulphur compounds, especially for the first synthesis of a polypeptide hormone…“ Pokud jde však o určení struktury, zvládl totéž v mnohem skromnějších technických a finančních podmínkách, než jaké měla k disposici du Vigneaudova skupina, ve stejné době mladý rakouský biochemik Hans Tuppy (*1924) spolu se svým tehdejším spolupracovníkem H. Michlem. Stalo se tak téměř paralelně s du Vigneaudem. Určil sekvenci aminokyselin oxytocinu u méně čistého oxytocinového preparátu chromatografickou metodoue [19,20]. Zásluha o určení struktury oxytocinu, což byl ve své době unikátní vědecký výkon, nepatří tedy výhradně du Vigneaudovi, ale i H. Tuppymu. Du Vigneaud tento fakt ve zmíněné přednášce při přebírání Nobelovy ceny přešel pouze bagatelizující poznámkouf [22].
V následujících létech uveřejnilo několik laboratoří alternativní metody syntézy oxytocinu, jejichž syntetická schémata shrnul do názorných diagramů R. A. Boissonas a St. Guttmann (Sandoz A. G., Basel) [23]. Jednou z nich byla elegantní syntéza vypracovaná v roce 1956 v Ústavu organické chemie a biochemie Československé akademie věd (dále také ÚOCHB) Josefem Rudingerem (1924–1975) a jeho spolupracovníky Janem Honzlem a Milanem Zaoralem [24,25]. Oproti du Vigneaudově syntéze se tato „pražská metoda“, spolu se syntézou oxytocinu z laboratoří Sandoz (R. A. Boissonnas a spol. [26]), projevila jako zvlášť metodicky výhodná a více uživatelsky praktická. „Stavebnicový“ postup, takzvané „trojkové“ schéma (obr. 1), pozůstávající ve spojování kratších peptidů (tripeptidů), navrhla jako alternativu k poněkud náročnému postupu du Vigneauda v následujících letech řada dalších autorů. Syntéza strukturních analogů neurohypofyzárních peptidů tím byla usnadněna a zrychlena. Diagram její verze použité Rudingerem a spolupracovníky je na obr. 1. Tato strategie ve farmakologii peptidů rovněž velice usnadnila studium vztahu závislosti mezi strukturou látky a jejími biologickými aktivitami. Endokrinologické charakteristiky, izolace a syntetické práce na obou hlavních hormonech neurohypofýzy – oxytocinu a arginin- či lysin-vasopresinu – probíhaly paralelně. I projekty syntézy analogů vycházely ze stejných či velmi podobných strategií. V době po publikaci prvních syntéz se jejich autoři převážně soustředili na izolované změny struktury jednotlivých pozic peptidového řetězce. Kombinace aminokyselin v jednom peptidovém řetězci vedly později k přípravě klinicky pozoruhodných farmak.

Strukturní analogy neurohypofyzárních peptidů a farmakologické strategie
Pražská „stavebnicová“ syntéza oxytocinu a strukturně obměněných molekul v oxytoxinu „v roztoku“ je v průmyslové výrobě používána často dodnes. Po zavedení pozdější a dnes široce používané syntézy v „pevné fázi“ Brucem R. Merrifieldem (1921–2006), který obdržel Nobelovu cena za chemii roku 1984, byla u některých léčivých látek tohoto typu zvolena pak tato metoda z hlediska správné výrobní praxe jako výhodnější. Problém dostupnosti a strategie volby analogů se později přesunuly do oblasti farmakologie a biochemie. Při studiu závislostí mezi strukturou a biologickou aktivitou (SAR – structure-activity relationships) neurohypofyzárních peptidů se v následujících „pionýrských“ letech jednotlivé výzkumné laboratoře zaměřily na různé cíle.
1. Vliv jednotlivých funkčních skupin či vazeb v peptidickém řetězci
„Intuitivní“ postup („trial-and-error“) použila především du Vigneaudova skupina. Cílem bylo zjistit vliv jednotlivých funkčních skupin či vazeb v peptidovém řetězci pomocí jejich modifikace či jejich vypuštěním. Tak byly získány první zajímavé analogy – jako příklad uveďme 2-O-methyltyrosin-oxytocin [27] a 2-D-tyrosin-oxytocin (substituce enantiomeru L-tyrosinu za D-formu). Především tak ale vznikly podklady pro další racionální postupy pro analýzu SAR.
2. Biologická aktivita jednotlivých analogů
Dalším cílem bylo studium spekter biologických aktivit jednotlivých analogů. Pro neurohypofyzární peptidy je typické, že vykazují pluripotentní aktivitu. Mají v různých proporcích uterokinetické, laktační, vazokonstrikční a antidiuretické účinky (a další, spíše farmakologicky zajímavé, účinky). Změna spektra aktivity biologických účinků se stala zájmem i základního výzkumu. Posun aktivit ve prospěch jedné z nich je často klinicky žádaný.
Dva příklady:
1) Analoga se zvýšeným poměrem vazokonstrikčního/antidiuretického účin- ku (analoga vasopresinu) syntetizovali R. A. Boissonnas, W. Doerpfner, St. Guttmann, K. Saameli a E. Stürmer ve výzkumných laboratořích firmy Sandoz v Basileji [28,29]. Klinicky významný byl svého času Octapressin® (2-fenylalanin-8-lysin-vasopresin), vykazující poměr vazokonstrikční/antidiuretická aktivita 2,5g, užívaný při lokální anestezii. Přidání této látky k lokálnímu anestetiku způsobilo vazokonstrikci a tím prodloužený anestetický účinek a nižší potřebu anestetika. Pro úpravu hemodynamiky, zejména u hemoragického šoku, byl navrhován 8-ornitin-vasopresin (Ornipressin), případně jeho analog (2-fenylalanin-8-ornitin) -vasopresin (poměr aktivit 4,0). Tyto látky, ve srovnání s jinými vazoaktivními aminy, byly výrazně výhodnější [30].
2) Mezi klinicky používanými analogy byl nejúspěšnější vasopresinový analog pražské skupiny, 1-deamino-8-D-arginin-vasopresin (DDAVP, Desmopressin, Minirin), syntetizovaný v laboratořích ÚOCHB (Milan Zaoral, Jaroslav Kolc, František Šorm) [31]. Extrémně zvýšená antidiuretická aktivita při minimální aktivitě vazokonstrikční tento analog předurčila být téměř výhradním lékem pro substituční terapii diabetes insipidus. Později bylo náhodně zjištěno, že tento analog zvyšuje hladinu koagulačního faktoru VIII jako hematostatikum při menších chirurgických (např. stomatologických) zásazích u osob s lehčí formou hemofilie a osob se sklonem k hemoragické diatéze.
3. Metabolická stabilita neurohypofyzárních hormonů
Specifickým cílem biochemické skupiny na ÚOCHB byla metabolická stabilita (inaktivace a clearance) neurohypofyzárních hormonů v souvislosti s jejich enzymovým odbouráváním in vivo. Tento záměr vedl chemiky k syntéze analogů s obměněnými místy potenciálních enzymových ataků v molekulách nativních hormonů. Vzniklo tak několik velice pozoruhodných analog (obr. 2).
Předně byl učiněn pokus o chránění peptidové vazby na N-konci peptidu, tedy mezi koncovým cysteinem a následujícím tyrosinem. Již v padesátých letech zjistili Tuppy a Nesvadba [32], že oxytocin je v průběhu těhotenství štěpen enzymem aminopeptidasového charakteru; později byl označen jako sérová či těhotenská oxytocinasa. Za částečně empiricky potvrzeného předpokladu, že aminopeptidasy tuto vazbu neštěpí, není-li aminoskupina přítomna, byl již du Vigneaudovou skupinou syntetizovaný – plně aktivní 1-deaminooxytocin rezistentní vůči enzymům tohoto typu. Později bylo, opět v laboratořích ÚOCHB, objeveno, že všechny oxytocinové a vasopresinové analogy s vypuštěnou či modifikovanou aminoskupinou skutečně vykazují prodloužené účinky. Podobná strategie byla použita i pro další místa možného enzymového štěpení, či vice versa k zodpovězení otázky, zda projektivní modifikace takových míst ve struktuře peptidu vede k prodloužení účinku a tím k identifikaci dalších inaktivujících enzymů. Tak se na příkladu dalo prokázat, že tyrosinasah [33], jiný možný inaktivační enzym neurohypofyzárních peptidů, je bez podstatného vlivu na kinetiku inaktivace: alkylace hydroxylové skupiny na druhé aminokyselině oxytocinu či vasopresinu délku účinku výrazně nezvyšuje. Protekční zásahy na karboxylovém, resp. karboxamidovém, konci řetězce nemají vliv na kinetiku biologických účinků, i když farmakologové z Mount Sinai University v New Yorku existenci enzymů štěpících oxytocin na tomto konci popsalii [34,35].
Disulfidový můstek mezi oběma cysteinovými řetězci v pozici 1 a 6 zaručuje aktivní cyklickou konformaci molekuly oxytocinu a s výjimkou zmíněné sérové oxytocinasy patrně i resistenci vůči aminopeptidasam. Redukce, resp. odstranění, disulfidického můstku vede k téměř neaktivnímu „lineárnímu“ oxytocinu. Zdá se, že specifická reduktasa oxytocinového můstku, která by vedla k inaktivaci tohoto hormonu, neexistuje, i když byla v biochemických laboratořích na ÚOCHB dlouho hledána. J. Rudinger a K. Jošt navrhli řešení otázky významu disulfidového můstku pro aktivitu a inaktivaci oxytocinu zcela unikátním způsobem. Navrhli náhradu jednoho nebo obou atomů síry neredukovatelnou „izosterní“ methylenovou skupinou (-CH2-) o zhruba stejném molekulárním objemu jako má atom síry (obr. 2). Tak vznikly pozoruhodné sloučeniny s biologickými aktivitami řádově srovnatelnými s nativní molekulou, označené jako „carba“-oxytociny [37]. Pod tímto jménem jsou dnes známy v chemickém a farmakologickém názvosloví mezinárodně. Kupodivu však nevykazují zvlášť prodloužené účinky, takže fyziologický význam inaktivace neurohypofyzárních peptidů redukcí disulfidického můstku byl tímto přinejmenším zpochybněn [38].
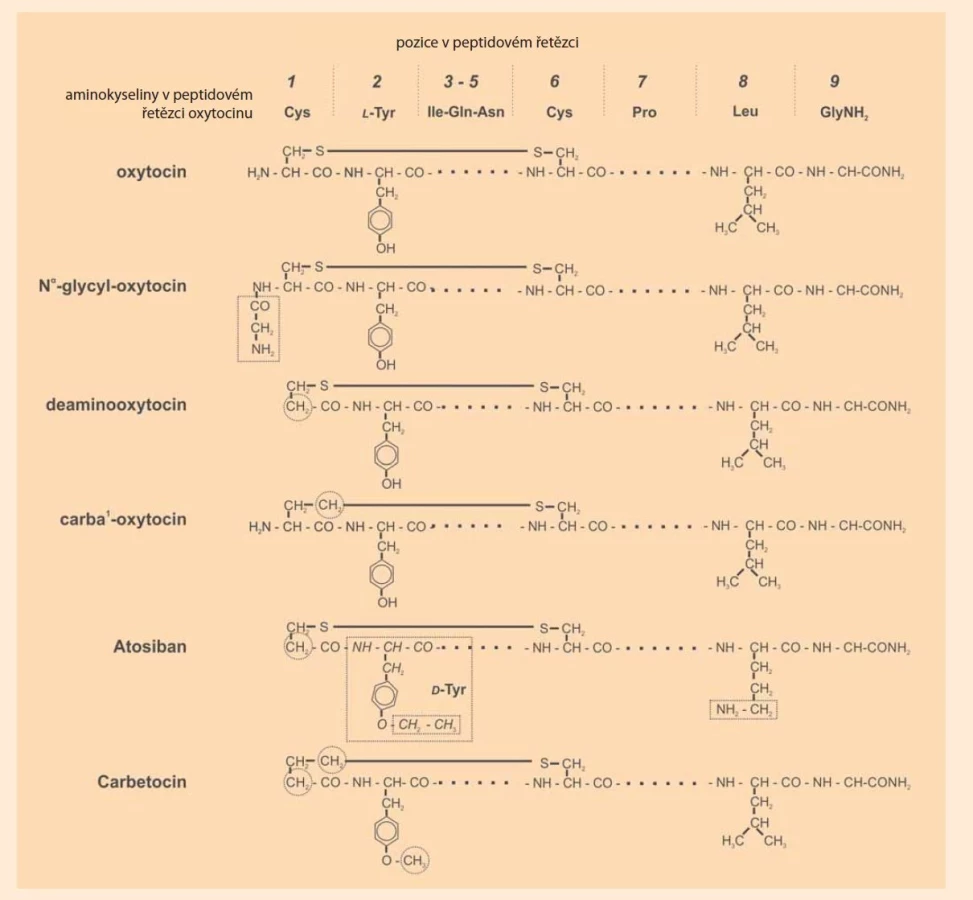
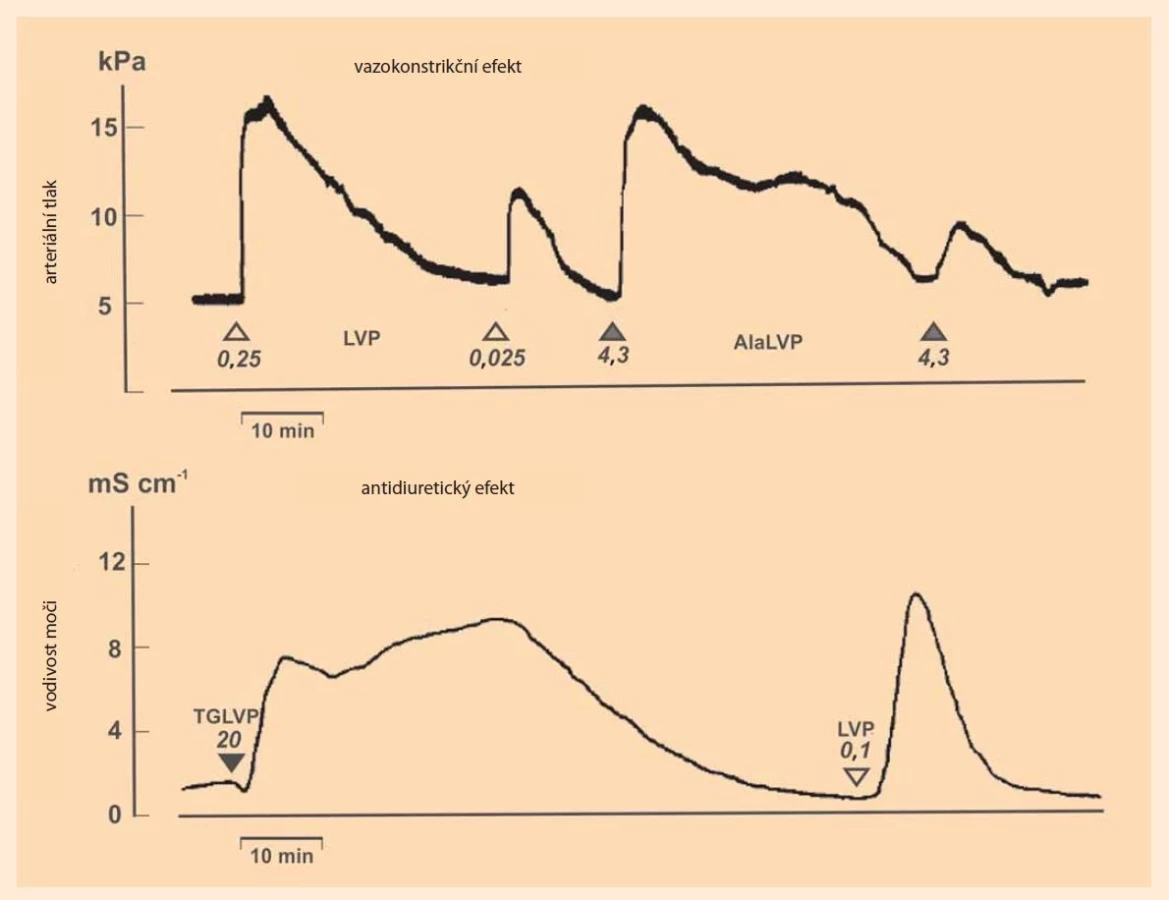
„Chráněné“ analogy umožnila nadto reverzní strategii tzv. enzymových sond (enzyme probes), tedy analýzu farmakokinetických procesů v blízkosti receptorů neurohypofyzárních peptidů, tedy v tzv. receptorovém kompartmentu [39]. Výsledky, stručně shrnuté, svědčí o tom, že stabilita peptidu, a tím i poločas účinku, souvisí převážně se strukturou aminokyseliny na jeho aminovém konci (na N-a-koncové aminokyselině). Kritický inaktivující enzym je tedy aminopeptidasa typu oxytocinasy popsané již výše zmíněnými autory Tuppym a Nesvadbou [32]. Snaha o prodloužení účinku, zvláště pro klinické využití, vedla k syntéze další významné skupině oxytocinových a vasopresinových analogů, k tzv. „syntetickým“ hormonogenům, u nichž byla na N-koncovou aminoskupinu připojena další aminokyselina či krátký peptid [40,41] (obr. 2). Předpokládalo se, že takto upravené molekuly se budou in vivo v prvém kroku enzymově štěpit aminopeptidasami, a teprve pak že dojde k uvolňování účinného hormonu. Farmakologické a imunologické podklady tento předpoklad u oxytocinových a lysin-vasopresinových hormonogenů plně potvrdily. Dva zvláště názorné příklady – časové profily antidiuretického a vazokonstrikčního účinku analogů lysin-vasopresinu – přináší obr. 3. Klinicky se uplatnil jako akutní prostředek při krvácivých stavech 1-N-a-triglycyl-8-lysin-vasopresin (TGLVP, Terlipressin, Glypressin), především v případě krvácení z jícnových varixů, a to kvůli svému vazokonstrikčnímu/hemostatickému účinkuj. O podobných oxytocinových analozích, především 1-N-a-triglycyl-oxytocinu a 1-N-a-leucyl-glycyl-glycyl-oxytocinu, existuje sice řada sdělení v literatuře, v gynekologicko-porodnické praxi se ale příliš neujaly. Výhody ve srovnání s oxytocinem se nezdají být výrazné.
4. Inhibice biologických aktivit neurohypofyzárních hormonů
Výzkum strukturních analogů působících jako inhibitory biologických aktivit neurohypofyzárních hormonů se datuje od rovněž spíše náhodného nálezu inhibice vazokonstrikčního účinku 8-arginin-vasopresinu 2-O-methyltyrosinoxytocinem na krysách. Jako první publikovali syntézu tohoto analogu v roce 1960 Law a du Vigneaud [27]; stalo se tak krátce před publikací stejné látky v laboratořích ÚOCHB [42]. Časový rozdíl obou publikací byl velmi krátký. Obě skupiny pracovaly na syntéze nezávisle a téměř současně. Pražská skupina však přesto v tomto případě ztratila primátk. Avšak brzy nato publikovali biochemici z ÚOCHB mnohem systematičtější studii inhibice uterotonického účinku oxytocinu tímto a dalšími analogy modifikovanými v pozicích aminokyselin 2, 3 a 4 (cit. [43], obr. 2). Popsané objevy dosvědčují, že tento typ strukturní modifikace byl na ÚOCHB již dříve ve středu zájmu. Z pražských výzkumných pracovišť pocházejí pak další studie těchto analogů, týkající se farmakologicky významného jevu, a to změny antagonismu v parciální a plný agonismus změnou pokusných podmínek [44]. Dodnes jsou vztahy mezi strukturou analogů a jejich inhibičními vlastnostmi celosvětově předmětem studií v četných laboratořích. Již na zlomu šedesátých a sedmdesátých let shrnul Josef Rudinger výsledky dosavadních studií v několika přehledech [45,46]. Přehled novějších prací, zaměřený převážně na význam antagonisticky působících analogů neurohypofyzárních peptidů a jejich možné léčebné využití, publikoval nedávno Maurice Manning (University of Toledo College of Medicine) se spolupracovníky z různých laboratoří [47].
5. Syntéza analogů s kombinacemi změn na různých pozicích peptidového řetězce
Období izolovaných obměn, které vedly k již známým změnám biologických aktivit, vystřídaly zhruba od 70. let pokusy s kombinacemi změn na různých pozicích peptidového řetězce, motivované převážně snahou připravit klinicky použitelná farmaka. K tomu dvě poznámky. První se týká kumulace strukturních změn v jednotlivých analozích. Tato nevede zpravidla ke kumulaci biologických vlastností spojených se stavem jednotlivých pozic v molekule. Vedla ovšem k syntéze řady pozoruhodných farmak. V případě oxytocinu takto vznikly dnes klinicky v porodnictví hojně používané léky carbetocin (Duratocin®/Pabal®) a (dnes již generikum) atosiban (Tractocile® a další generické názvy jeho lékových forem).
Poznámka druhá, biologický fenomén charakterizovaný farmakologicky se nemusí nutně v podobné formě projevit klinicky. Výrazně se to týká farmakokinetických vlastností konkrétních látek, když poločasy účinků měřených v pokusech ex vivo se liší od pozorování in vivo. V klinické praxi používaný analog vasopresinu, 1-deamino-8-D-arginin-vasopresin (Desmopressin, DDAVP), vykazoval v antidiuretickém testu při aplikaci ekvipotentních dávek odpovídajících 8-arginin-vasopresinu jen mírně prodloužený poločas (nejspíše vlivem vypuštění aminoskupiny na N-konci), ale téměř dramaticky prodloužený účinek při intranazálním podání pacientům s diabetes insipidus. Příčinou ovšem může být i vznik depotních forem, například vazbou peptidu ve velké dávce na bílkoviny a jeho postupného uvolňování. Podobný efekt lze pozorovat u carbetocinu, 1-deamino-carbal-2-O-methyltyrosin-oxytocinu. Carbetocin se v dnešní době používá v porodnictví pro prevenci a/nebo léčbu peripartálního život ohrožujícího krvácení. Jak již bylo zmíněno, samotná substituce –S– atomu skupinou –CH2– neprodlužuje poločas rozpadu oxytocinu, avšak v klinické situaci výrazně prodlouženě působí. Klinický význam carbetocinu pak spočívá v uterotonickém účinku. Oproti oxytocinu navozuje silnější a dlouhodobější retrakci myometria při porodu placenty člověka, tedy ve III. době porodní. Proto je vedle preventivního podání používán i léčebně, a to při poporodní hypotonii a/nebo atonii hladké svaloviny dělohy.
Dva protagonisté ve výzkumu neurohypofyzárních peptidů v Praze
Od poloviny 50. do konce 60. let minulého století byl český výzkum obou hormonů neurohypofýzy zcela nerozlučně spojen s osobnostmi Josefa Rudingera a Františka Šorma (1913–1980) (obr. 4). První z nich byl už od počátku své akademické kariéry především světově uznávaný chemik v oblasti peptidů. Byl spíše konstruktivní teoretik než experimentátor. Kvůli svému neobyčejnému jazykovému nadání byl současně vynikající organizátor; dovedl pohotově shrnout pohledy odborníků různých oborů a formulovat z nich vědecké závěry. Byl proto také vítaným hostem na nejrůznějších vědeckých shromážděních. Jako první přišel s myšlenkou inovativní syntézy oxytocinu v Ústavu organické chemie a biochemie Československé akademie věd. Avšak vzhledem ke své rodinné historii (otec odsouzen v procesech na počátku 50. let k dlouhému trestu odnětí svobody) by své myšlenky v raných létech komunistické diktatury neuskutečnil bez jednoznačné podpory druhého jmenovaného, profesora Františka Šorma, tehdejšího ředitele ÚOCHB a pozdějšího prezidenta Československé akademie věd.

Několik slov k F. Šormovi. Byl přesvědčený (snad?) komunista, člen ústředního výboru KSČ, v době komunistického režimu poslanec Národního shromáždění. V tehdejším totalitním státě se tedy jednalo o pevně zakotvenou osobnost. V prvé řadě byl ale profesor a vědec. Jednal přímo, energicky a svoje tehdejší postavení nezneužíval v neprospěch vědy a sotva kdy k vlastnímu prospěchu. „Kádrové“ záležitosti ho nezajímaly, kritériem pro výběr spolupracovníků byl, snad jen s malými výjimkami, vědecký talent. Tím lze i vysvětlit, že jedním z prvních jím vybraných spolupracovníků byl Josef Rudinger, a to i přes jeho již zmíněnou rodinnou historii. Obsáhlý životopis Josefa Rudingera publikoval jako editorial v roce 2004 John Jones [48].
Zásluhou obou byla založena tradice dodnes se rozvíjejících Evropských peptidových sympozií*: V době, kdy komunistický režim naprosto zabraňoval stykům se Západem, se právě první Evropské peptidové sympozium konalo v roce 1958 v Praze. Toto svým stranickým vlivem, z podnětu Josefa Rudingera, umožnil František Šorm. Snad nejlépe bude tento počin charakterizovat pasáž z knihy The World of Peptides dvou významných chemiků, Theodora Wielanda a Miklose Bodanszkyho, spolupracovníka Vincenta du Vigneauda [49]: „We cannot enumerate here Rudinger’s many valuable contributions to the methodology of peptide synthesis or to the design of molecules for specific pharmacological studies. It seems to be more important to recall one of his ideas, that had and still has a major impact on peptide chemistry. In 1958 he invited the handful of European investigators active in peptide research to participate in a symposium in Prague. This first European Peptide Symposium was followed […] by meetings held in various locations in Europe. Their excellence acted as a stimulus or perhaps as catalyst and led to an unprecendent growth in research and understanding of peptide chemistry. Not less important are the friendships and collaborations formed at the Symposia. Their example was followed in the United States and also Japan …“
Po sovětské okupaci, již koncem srpna 1968, odešel Josef Rudinger z Československa do Švýcarska. Jako světoznámý vědec se stal na jedné z nejlepších evropských univerzit, Eidgenössische Technische Hochschule (ETH) v Zürichu, jedním z prvních profesorů molekulární biologie. Věnoval se, vedle četných dalších úkolů spadajících do povinností vysokoškolského profesora, spolu se svojí skupinou až do své předčasné smrti v roce 1975 nadále chemii a biologii peptidů. Avšak vědecky nejproduktivnější bylo bezesporu jeho životní období na pražském Ústavu organické chemie a biochemie v Praze. Výzkum peptidů ovšem na tomto ústavu pokračoval dále, přesto že Rudingerova nepřítomnost zanechala nepřekonatelnou mezeru. Jeho roli převzalo několik vynikajících chemiků, z nich je na místě zmínit alespoň Karla Jošta (1932–1986), Karla Podušku (†1974, datum neověřeno) a Milana Zaorala (1926–2011) (obr. 5). První dva rovněž předčasně zemřeli, ve stejné době jako Josef Rudingerm. M. Zaoral se pak od původní „peptidové skupiny“, vedené po odchodu Josefa Rudingera Karlem Bláhou (1926–1987), odpojil a vedl vlastní nezávislou laboratoř.

Oxytocin a vasopresin v laboratořích a na klinikách v Praze
Dostupností analog, které pocházely z prací Rudingerovy skupiny, iniciovala pozdější farmakologický, biochemický a klinický výzkum neurohypofyzárních hormonů na různých pracovištích v Praze. V 60. letech existovala již v Praze, zásluhou Františka Šorma, rozsáhlá komunikace se západními a středoevropskými pracovišti, především v Maďarsku, v Polsku, v NDR, ve Švýcarsku, ve Velká Británii a v Německé spolkové republice. I v této době již existovala mezinárodní výměna informací a často i preparátů. Centrální osobou byl v úloze jakéhosi vyslance opět Josef Rudinger. V té době se stal pražský výzkum peptidů součástí světového výzkumu.
Biochemický a zčásti též farmakologický výzkum byl na Ústavu organické chemie a biochemie soustředěn v oddělení molekulární biologie, zpočátku v laboratoři vedené Ivanem Rychlíkem (Ivan Rychlík, Zdeňka Beránková-Ksandrová, Ivan Bartošek, Vladimír Pliška, Tomislav Barth, Tomáš Douša), poslední tři jmenovaní spolu s dalšími spolupracovníky v oddělené laboratoři biochemie a farmakologie peptidů. Kromě metabolických a farmakokinetických studií byl zájem zaměřen na antidiuretické a natriferické účinky analogů vasopresinu a oxytocinu. Farmakokinetická charakteristika peptidů a originální antidiuretický test na kryse pocházející z této laboratoře se rozšířily ve farmakologickém výzkumu a farmaceutickém průmyslu [50].
Z ÚOCHB rovněž pocházejí jedny z prvních konformačních studií oxytocinu a jeho carba-analogů (Ivo Frič, Milan Kodíček, Karel Jošt, Karel Bláha [51]).
Velkou část farmakologického výzkumu převzal pražský Výzkumný ústav přírodních léčiv (Ivan Krejčí, Ivo Poláček, Běla Kupková, Alena Machová a řada dalších). Odtud pochází většina dat k uterotonickým a vazokonstrikčním aktivitám peptidů z ÚOCHB. Ivan Krejčí se spolupracovníky se nadto zabýval základními otázkami farmakologie peptidů. Studovali inhibici uterotonického efektu oxytocinu, přechody mezi plným agonismem a antagonismem, a vlivem vápníku a hořčíku na tyto procesy. Jeho skupina přešla koncem 60. let do Výzkumného ústavu pro farmacii a biochemii. I tam přetrvala tematika oxytocinových analog 70. léta a končila až v novém tisíciletí. Rovněž, avšak snad v menší míře, spolupracovali v tomto ústavu s biochemickou skupinou ÚOCHB Karel Řežábek, Evžen Kasafírek a Jaroslav Kynčl na syntéze, především však na farmakologické charakteristice vazokonstrikčních hormonogenů.
Projekty z oboru kardiovaskulární farmakologie a fyziologie vasopresinu byly až do počátku 70. let součástmi programu Ústavu pro choroby oběhu krevního v Praze-Krči (Jiří Heller, nějaký čas také Joseph H. Cortn).
To je ovšem pouze stručný výčet pražských farmakologických a fyziologických pracovišť, která se na výzkumu „pražských“ analog oxytocinu a vasopresinu podílela. Do tohoto proudu byla zapojena i řada pražských klinických zařízení, především z oboru gynekologie a porodnictví. Zde patří zmínit Ústav pro péči o matku a dítě (Vladimír Brotánek, Stanislav Kazda, J. Hodr, Č. Jungmannová, J. Židovský) a Gynekologicko-porodnická klinika fakulty dětského lékařství Univerzity Karlovy, dnes Gynekologicko-porodnická klinika 2. LF UK a Fakultní nemocnice v Motole. V té době byl přednostou Alfréd Kotásek, po něm Miroslav Břešťák. Farmakologický a klinický výzkum byl také předmětem endokrinologické laboratoře III. interní kliniky Všeobecné fakultní nemocnice v Praze pod vedením profesorů Vladimíra Holečka, Josefa Marka a Vratislava Schreibera.
V roce 1968 započala spolupráce se švédskou firmou Ferring Läkemedel AB, Malmö. Pokračovala pak prodejem licence na dDAVP/Desmopressin (později i prodejem dalších patentů), čímž tato společnost získala právo vyrábět Desmopressin, Carbetocin/Pabal a Terlipressin v západní Evropě a USA. Firma Léčiva, n. p., jako součást trustu Spofa pak vyráběla v jednom ze svých závodů v Praze-Komořanech stejné peptidy pro státy východní Evropy a Sovětský svaz.
Pražský výzkum později změnil svůj charakter a orientoval se více na klinickou farmakologii. Řada chemickosyntetických a farmakologických projektů se pak soustředila nejprve v Malmö, později v dalších pobočkách této rychle rostoucí a zvětšující se firmy ve světě. Pražský „Gründerzeit“ má zde svůj přirozený, přesto velmi úspěšný konec.
Autoři jsou zavázáni díky paní Mgr. Olze Štajnrtové z Gynekologicko-porodnické kliniky 1. LF UK a VFN v Praze za pečlivé jazykové a věcné korektury rukopisu, zvláště pak za ověření a opravu citovaných údajů.
a Citát v Lancet1 (s. 340) „…These chemical messengers […] or,hormones’ (from όρμάω, I excite or arouse), as we might call them, have to be carried from the organ where they are produced to the organ which they affect by means of the blood stream …“
b Bayliss a Starling docházejí v tomto sdělení k opačným závěrům než Ivan Petrovič Pavlov (1849–1936), který deklaroval přímou a výhradní roli nervového impulsu a přítomnosti chymu v duodenu na pankreatickou sekreci. Jeho nálezy diskutují kontroverzně a poukazují na existenci humorální regulace. Příspěvek Pavlova (Nobelova cena za fyziologii či medicínu v roce 1904, tedy dva roky po publikaci jejich výsledků) citují autoři se značným, plně kolegiálním respektem a oceněním. Nicméně nadepisují příslušnou kapitolu ve své publikaci titulem VII. The normal mechanism, chemical or nervous?
c Organoterapií se označovalo používání tkáňových extraktů k terapeutickým účelům.
d Britský fysiolog, později profesor fyziologie (University of Manitoba in Winnipeg, Kanada).
e Hans Tuppy měl již v tomto oboru nejen zkušenosti, ale i významné výsledky: Na roční stáži v roce 1949 určil v laboratoři F. Sangera v Cambridge strukturu B-řetězce inzulinu. Frederick Sanger jeho podstatný příspěvek ke struktuře inzulinu s uznáním zdůraznil ve své prvé nobelovské přednášce [21] (cena za chemii 1958): „… the work on fraction B progressed so favourably and Tuppy worked so hard that by the end of the year we were virtually able to deduce the whole of the sequence of its 30 residues.“
f Cit. 22: „It is of considerable interest that Tuppy, on the basis of data we had published along with some data of his own, arrived at the same structure independently. Tuppy’s proposal was based on the data from our laboratory on composition, molecular weight, terminal groups, as worked out by our use of the Sanger dinitrophenyl end group procedure …“
g Test na kryse. Poměr aktivit pro arginin-vasopresin (standard) je 1,0.
h Enzym katalyzující oxidaci fenolů v ortho-pozici k hydroxylové skupině tyrosinu (oxytyrosinasa), či – za přítomnosti exogeního kyslíku – na chinony. Lze předpokládat, že takto modifikovaná molekula oxytocinu je biologicky neaktivní.
i Peptidy vzniklé odštěpením koncového glycinamidu zmíněnými enzymy (karboxamidopeptidasami) – zvláště desglycinamid-8-lysin-vasopresin – mohou mít četné účinky na různá mozková centra [36]. Dlouhá léta se jimi zabýval David de Wied (1925–2004) se spolupracovníky (zvláště B. Bohus, T. B. van Wimersma Greidanus) na univerzitě v Utrechtu (Rudolf Magnus Institut). Jde ovšem o účinky sekundární; jejich regulační fyziologická funkce není dostatečně doložena.
j Podle neověřených sdělení byl jako akutní protišokový prostředek zajímavý pro sanitní sbory některých armád. Další testovanou farmakologickou možností bylo použití Terlipressinu jako abortiva v prvním trimestru.
k Jeden z autorů tohoto sdělení (V. P.) si vzpomíná, že Josef Rudinger se ihned po uveřejnění syntézy skupinou du Vigneauda vyjádřil, že na primátu nezáleží, hlavně že analog byl syntetizován. To je přesvědčivý příklad vysoké vědecké morálky Josefa Rudingera.
l Jsou do dnešního dne organizovány prostřednictvím European Peptide Society vždy v sudých letech v jednotlivých evropských státech.
m V 70. letech zemřelo několik peptidových chemiků v Praze a v zahraničí (Itálie, USA) před dosažením padesáti let nebo krátce po padesátce, nejméně tři z pěti nám známých s onkologickou diagnózou. Podle nepotvrzené domněnky mohlo jejich onemocnění souviset s dlouhodobou expozicí dicyclohexylcarbodiimidem (DCC), běžně používaného v chemii peptidů jako kondenzační činidlo. Jeho kancerogenitu deklaruje National Institute of Health (NIH Publication No. 07-4426).
n Poznámka k osobě Josepha H. Corta, jehož jméno se často objevuje mezi autory pražských publikací v 60. a 70. letech. Cort emigroval v 50. letech z USA, údajně před hrozbou žaloby ze strany komise McCarthyho, do Anglie a brzy poté nejprve do Polska a pak do Československa. Jako údajná oběť pronásledování komunistů na Západě byl oficiální Prahou vítán. (Nutno podotknout, že před kolegy v Praze používal zcela jiný, vůči komunismu satiricko-kritický postoj.) Jako absolvent Yale University se připojil k výzkumným pracím Josefa Rudingera, s kterým se úzce přátelil. Mimo zmíněný ústav je v publikacích uváděn jako jeho pracoviště též Fyziologický ústav ČSAV v Krči (laboratoř biologie peptidů). Byl bezesporu nadaným experimentátorem, měl řadu originálních návrhů, hlavně se týkaly vlivu neurohypofyzárních hormonů na iontový metabolismus. O jeho morálním profilu, alespoň pokud šlo o vědu, existovaly již tehdy v zasvěcených kruzích značné pochyby. Mimo jiné byly výsledky jeho pokusů často nereprodukovatelné. V roce 1976 se vrátil do Spojených států, kde působil nejprve na Mount Sinai Medical Center v New Yorku, později ve firmě Vega Biotechnologies, Tucson (Arizona). Koncem roku 1982 veřejně přiznal, že jeden z analogů vasopresinu, který údajně farmakologicky testoval a na který mu byl udělen americký patent, ve skutečnosti nebyl nikdy syntetizován. O dalších jím deklarovaných podobných peptidech rovněž neexistují doklady, zda vůbec kdy syntetizovány byly. Tento vyslovený podvod okamžitě vzbudil veřejný skandál a zřejmě ukončil Cortovu kariéru (viz např. New Scientist, January 6, 1983, New York Times, December 27, 1982 atd.). Naštěstí nevrhla tato historie dlouhé stíny na práci pražských výzkumníků. Pokud víme, Cort si pak otevřel firmu ve Wiesbadenu, kde se pokoušel mimo jiné vyrábět také carbetocin.
Poděkování časopisu Chemické listy za možnost publikování.
Zdroje
1. Starling E. H.: Lancet 2, 339 (1905).
2. Bayliss W. M., Starling E. H.: J. Physiol. 28, 325 (1902).
3. von Mering J., Minkowski O.: Arch. Exp. Path. Pharm. 26, 371 (1890).
4. Oliver G., Schäfer E. A.: J. Physiol. 18, 230 (1895).
5. Oliver G., Schäfer E. A.: J. Physiol. 18, 277 (1895).
6. Howell W. H.: J. Exp. Med. 3, 245 (1898).
7. Schäfer E. A., Vincent S.: J. Physiol. 25, 87 (1899).
8. Dale H. H.: J. Physiol. 34, 163 (1906).
9. Ott I., Scott J. C.: Proc. Soc. Exp. Biol. Med. 8, 48 (1910).
10. Dudley H. W.: J. Pharmacol. Exp. Ther. 14, 295 (1919).
11. Pliška V., Barth T.: Collect. Czech. Chem. Commun. 35, 1576 (1970).
12. Kamm O., Aldrich T. B., Grote I. W., Rowe L. W., Bugbee E. P.: J. Am. Chem. Soc. 50, 573 (1928).
13. Stehle R. L., Fraser A. M.: J. Pharmacol. Exp. Ther. 55, 136 (1935).
14. Craig L. C.: J. Biol. Chem. 155, 519 (1944).
15. Livermore A. H., du Vigneaud V.: J. Biol. Chem. 180, 365 (1949).
16. Edman P.: Arch. Biochem. 22, 475 (1949).
17. du Vigneaud V., Ressler C., Trippett S.: J. Biol. Chem. 205, 949 (1953).
18. du Vigneaud V., Ressler C., Swan J. M., Roberts C. W., Katsoyannis P. G.: J. Am. Chem. Soc. 76, 3115 (1954).
19. Tuppy H.: Biochim. Biophys. Acta 11, 449 (1953).
20. Tuppy H., Michl H.: Monatsh. Chem. 84, 1011 (1953).
21. Sanger F., v knize: Nobel lectures – chemistry 1942–1962. Including presentation speeches and laureates’ biographies, str. 544. Nobel Foundation by Elsevier Publishing Company, Amsterdam 1964.
22. du Vigneaud V., v knize: Nobel lectures – chemistry 1942–1962. Including presentation speeches and laureates’ biographies, str. 446. Nobel Foundation by Elsevier Publishing Company, Amsterdam 1964.
23. Boissonnas R. A., Guttmann S., v knize: Neurohypophysial Hormones and Similar Polypeptides (B. Berde, ed.), kap. 2, str. 40. Springer Verlag, Berlin 1968.
24. Rudinger J., Honzl J., Zaoral M.: Collect. Czech. Chem. Commun. 21, 202 (1956).
25. Honzl J., Rudinger J.: Collect. Czech. Chem. Commun. 20, 1190 (1955).
26. Boissonnas R. A., Guttmann S., Jaquenoud P.-A., Waller J.-P.: Helv. Chim. Acta 28, 1491 (1955).
27. Law, H. D., du Vigneaud V.: J. Am. Chem. Soc. 82, 4579 (1960).
28. Berde B., v knize: Neurohypophysial Hormones and Similar Polypeptides (B. Berde, ed.), kap. 16, str. 802. Springer Verlag, Berlin 1968.
29. Jošt K., v knize: Handbook of Neurohypophyseal Hormone Analogs, Vol. II/2 (K. Jošt, M. Lebl, F. Brtník ed.), str. 94. CRC Press, Inc., Boca Raton (FL) 1987.
30. Altura B. M.: J. Pharmacol. Exp. Ther. 198, 187 (1976).
31. Zaoral M., Kolc J., Šorm F.: Collect. Czech. Chem. Commun. 32, 1250 (1967).
32. Tuppy H., Nesvadba H.: Monatsh. Chem. 88, 977 (1957).
33. Chang T.-S.: Int. J. Mol. Sci. 10, 2440 (2009).
34. Walter R., Havran R. T., Schwartz I. L.: J. Med. Chem. 19, 328 (1976).
35. Walter R., Schlank H., Glass J. D., Schwartz I. L., Kerenyi T. D.: Science 173, 827 (1971).
36. van Wimersma Greidanus T. B., Van Ree J. M., v knize: Behavioral Aspects of Neuroendocrinology (D. Ganten, D. Pfaff, ed.), kap. 3, str. 61. Springer Verlag, Berlin 1990.
37. Rudinger J., Jošt K.: Experientia 20, 570 (1964).
38. Pliška V., Rudinger J., Douša T., Cort J. H.: Am. J. Physiol. 215, 916 (1968).
39. Pliška V., Jutz G., Beck S., v knize: PEPTIDES: Structure and Function. Proceedings of the Ninth American Peptide Symposium (C. M. Deber, V. J. Hruby, K. D. Kopple, ed.), str. 603. Pierce Chemical Company, Tucson (AR) 1985.
40. Beranková-Ksandrová Z., Bisset G. W., Jošt K., Krejčí I., Pliška V., Rudinger J., Rychlík I., Šorm F.: Brit. J. Pharmacol. 26, 615 (1966).
41. Kynčl J., Řežábek K., Kasafirek E., Pliška V., Rudinger J.: Eur. J. Pharmacol. 28, 294 (1974).
42. Jošt K., Rudinger J., Šorm F.: Collect. Czech. Chem. Commun. 26, 2496 (1961).
43. Beránková Z., Rychlík I., Jošt K., Rudinger J., Šorm F.: Coll. Czech. Chem. Commun. 26, 2673 (1961).
44. Krejčí I., Kupková B., Rudinger J.: Brit. J. Pharmacol. 30, 506 (1967).
45. Rudinger J., Krejčí I., v knize: Neurohypophysial Hormones and Similar Polypeptides (B. Berde, ed.), kap. 15, str. 748. Springer Verlag, Berlin 1968.
46. Rudinger J., Pliška V., Krejčí I.: Recent Prog. Horm. Res. 28, 131 (1972).
47. Manning M., Misicka A., Olma A., Bankowski K., Stoev S., Chini B., Durroux T., Mouillac B., Corbani M., Guillon G.: J. Neuroendocrinol. 24, 609 (2012).
48. Jones J.: J. Pept. Sci. 10, 393 (2004).
49. Wieland T., Bodanszky M.: The World of Peptides. A Brief History of Peptide Chemistry, str. 156. Springer Verlag, Berlin 1991.
50. Thorn N. A. v knize: Neurohypophysial Hormones and Similar Polypeptides (B. Berde, ed.), kap. 7, str. 372. Springer Verlag, Berlin 1968.
51. Frič I., Kodíček M., Jošt K., Bláha K.: Collect. Czech. Chem. Commun. 39, 1271 (1974).
Štítky
Dětská gynekologie Gynekologie a porodnictví Reprodukční medicínaČlánek vyšel v časopise
Česká gynekologie

2023 Číslo 1
Nejčtenější v tomto čísle
- Kryokonzervace ovariální tkáně jako metoda pro zachování fertility u žen
- Předčasný odtok plodové vody před termínem porodu
- Fertilitu šetřící terapie u ektopické gravidity
- Staré a nové pohľady na funkčnú morfológiu vajíčkovodov a ich význam pre gynekologickú prax