
Myomatóza děložních rudimentů u pacientky s Mayer-Rokitansky-Küster-Hauser syndromem
Myomas in uterine rudiments in a patient with Mayer-Rokitansky-Küster-Hauser syndrome
Objective: To describe the case of a patient with Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome and a history of chronic pelvic pain due to myomas in the rudimentary uterine horns. The article highlights a rare origin of gynaecological pain.
Case report: We present the case of 61-year-old woman with MRKH syndrome who has suffered from chronic abdominal pain for more than one year before surgery. Using magnetic resonance imaging and ultrasonography, a suspicion on the tumours of uterine myoma character in the rudimentary horns was suggested. It was confirmed by laparoscopy. Myomas were removed in the endobag and histopathologically confirmed.
Conclusion: MRKH syndrome is a very rare disease. Approximately ten women are annually born with this congenital anomaly in the Czech Republic. While myoma incidence is extremely rare in this group of women, it must be taken into account in differential diagnosis and solved surgically in time.
Keywords:
Mayer-Rokitansky-Küster-Hauser syndrome – rudimentary uterine horn – myoma uteri
Authors:
Hricko M. 1; Chmel R. jr. 1,2; Pastor Z. 1
Authors‘ workplace:
Gynekologicko-porodniká klinika, 2. LF UK a FN Motol, Praha 2 LF UK v Plzni
1
Published in:
Ceska Gynekol 2021; 86(1): 36-39
Category:
Case Report
doi:
https://doi.org/10.48095/cccg202136
Overview
Cíl práce: Popis případu pacientky s Mayer-Rokitansky-Küster-Hauser (MRKH) syndromem a anamnézou chronické pánevní bolesti způsobené rostoucími myomy vycházejícími z rudimentárních rohů dělohy. Článek upozorňuje na vzácnou příčinu bolesti v podbřišku způsobenou gynekologickým onemocněním.
Kazuistika: Předkládáme případ 61leté ženy s MRKH syndromem, která více než 1 rok trpěla chronickými bolestmi břicha. Pomocí magnetické rezonance a ultrasonografie bylo vysloveno podezření na tumory charakteru děložních myomů, které byly laparoskopicky odstraněny a histopatologicky potvrzeny.
Závěr: MRKH syndrom je velmi vzácný. V ČR se narodí přibližně deset žen s touto diagnózou ročně. Výskyt myomů je u této skupiny žen sice extrémně raritní, ale je nutné ho vzít při diferenciální diagnostice v úvahu a včas chirurgicky řešit.
Klíčová slova:
Mayer-Rokitansky-Küster-Hauser syndrom – rudimentární roh dělohy – myoma uteri
Úvod
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrom je charakterizován aplazií dělohy a horních dvou třetin pochvy. Může být spojen s renálními, skeletálními, sluchovými a srdečními malformacemi. Jeho prevalence se odhaduje na 1: 4 000–5 000 narozených žen [1]. Vychází z vývojového postižení Müllerových vývodů neznámé etiologie v průběhu organogeneze. V současnosti neznáme jasnou genetickou příčinu MRKH syndromu [2], nicméně v některých případech dochází k jeho výskytu v širším příbuzenstvu, a proto lze předpokládat nějakou formu jeho genetické i epigenetické determinace [3]. Fenotyp těchto žen je jednoznačně ženský, karyotyp je 46 XX, mají fyziologické endokrinní funkce, bifázický ovariální cyklus a ženskou psychosexuální identifikaci [4]. Rozlišujeme tři formy tohoto syndromu:
a) typická forma s absencí dělohy a horní vaginy s normálními vaječníky a vejcovody;
b) atypická forma syndromu MRKH s dalšími orgánovými malformacemi (např. ageneze ledviny);
c) uterovaginální aplazie/hypoplazie spolu s jinými vrozenými anomáliemi (renální, skeletální, sluchové, srdeční a oční), je označována také jako MURCS (Müllerian duct aplasia-renal agenesis-cervicothoracic somite dysplasia) syndrom [5,6].
U části pacientek se syndromem MRKH nacházíme pozůstatky rudimentárních děložních struktur obsahujících buňky myometria (až v 80 % případů) [7], které mohou být příčinou vzniku děložních myomů. První případ leiomyomu dělohy u ženy se syndromem MRKH byl popsán v roce 1977 [8,9]. V naší kazuistice chceme upozornit na neobvyklou etiologii chronických bolestí břicha, která může být v některých případech příčinou náhlé příhody břišní a je nutné s ní při diferenciální diagnostice počítat.
Kazuistika
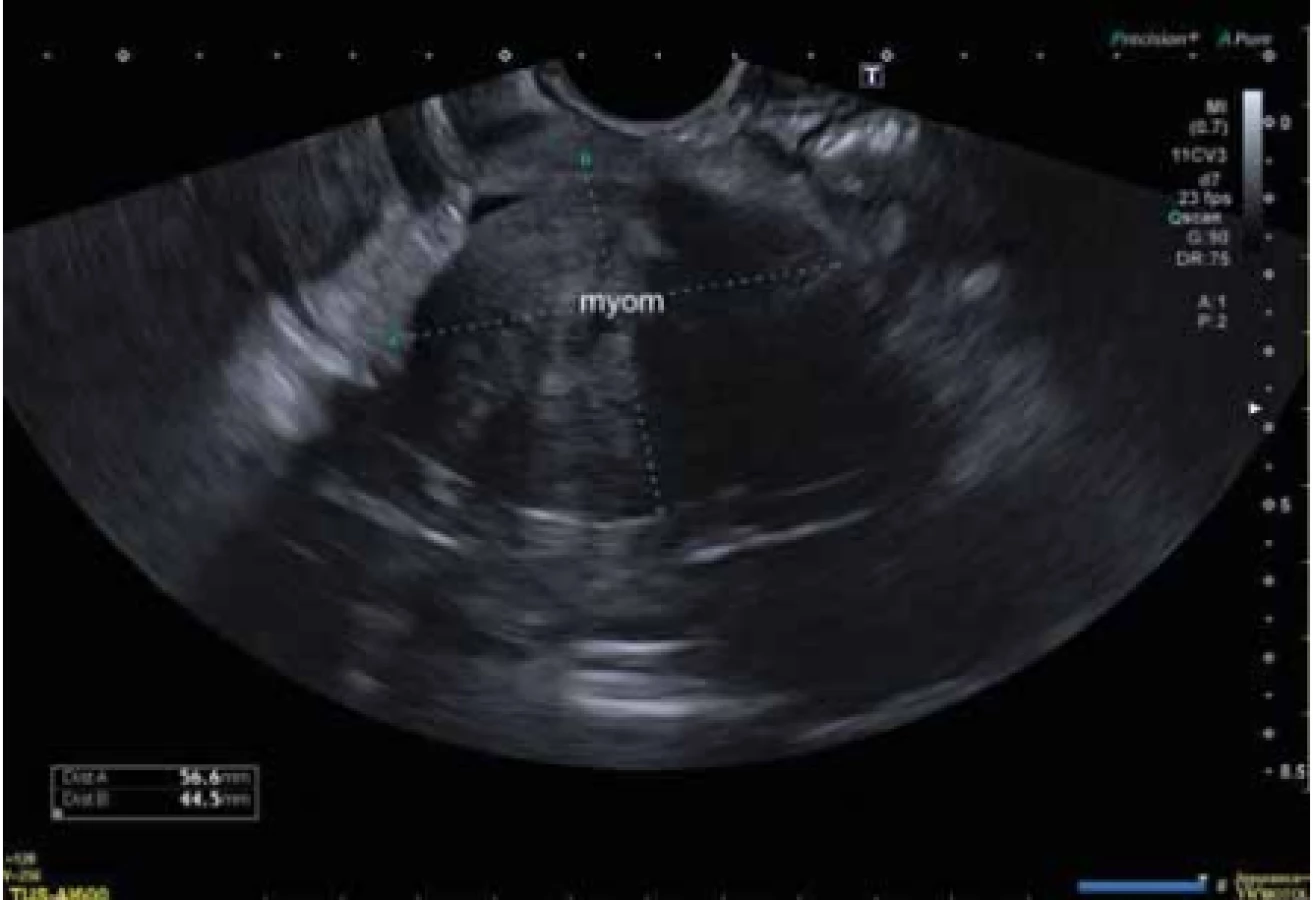
Na naší klinice byla vyšetřena 61letá pacientka, u které byl v 16 letech diagnostikován MRKH syndrom. V posledním roce si stěžovala na pocit vzedmutého či plného podbřišku (bulk symptoms), 2 měsíce před operací udávala intermitentní bolesti v epigastriu. V 18 letech podstoupila nechirurgickou dilatační terapii k vytvoření neovaginy. Za posledních 24 let nebyla gynekologicky vyšetřena. Jednalo se o zdravou ženu bez rizikové anamnézy, nekuřačku s body mass indexem (BMI) 28 kg/m2 a bez jakékoli medikamentózní terapie. Její pochva byla ze dvou třetin obliterována, délky 30 mm, palpační bimanuální vaginální vyšetření bylo proto velmi limitované. Nebyly přítomny známky peritoneálního dráždění. Pomocí ultrasonografického vyšetření byl diagnostikován nehomogenní hypoechogenní tumor malé pánve velikosti 56 × 44 × 74 mm (obr. 1).
Fig. 1. Ultrasound examination before surgery, hypoechogenic tumor of the small
pelvis, size 56 × 44 × 74 mm.
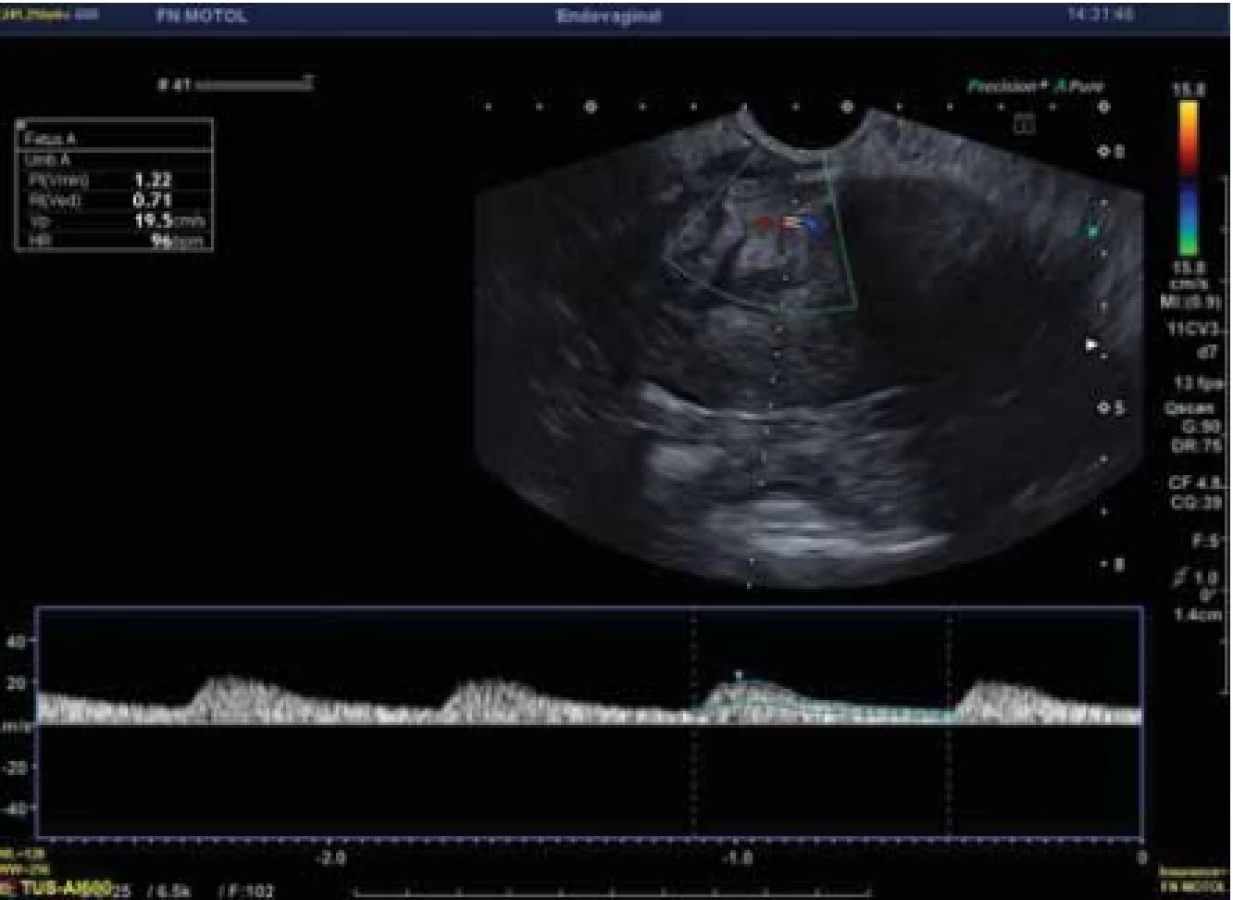
Dopplerovská flowmetrie vykazovala nízkou až střední perfuzi s nízkoodporovými toky (PI 1,2), color score (CS) bylo 2 (obr. 2).
Fig. 2. Doppler flowmetry of the investigated structure.
Ovaria byla normální velikosti a struktury uložené kraniolaterárně, výše než obvykle. Biochemické onkogenní (CEA, CA 125) a zánětlivé (CRP, prokalcitonin) markery byly negativní. Magnetická rezonance s kontrastem odhalila dobře ohraničený útvar v místě rudimentu rohu dělohy velikosti 80 × 48 × 47 mm, bez restrikce perfuze s postkontrastně výrazněji nasycenou periferií. Obdobná, ale menší ložiska byla popsána i dorzolaterálně vpravo (30 × 20 × 20 mm) a ventrolaterálně vlevo (12 × 6 × 7 mm) od největšího tumoru (obr. 3, 4). Při laparoskopii byl verifikován nález tří myomů vycházejících z rudimentárních děložních rohů. Největší myom vycházel zleva, byl torkvovaný o 360°, bez vizuálních trofických změn a v pravém děložním rudimentu byly nalezeny další myomy o velikosti 30 a 15 mm. Všechny myomy byly exstirpovány, největší z nich byl extrahován po jeho částečné morselaci v endobagu (obr. 5).
Fig. 3. Magnetic resonance imaging findings – axial view.
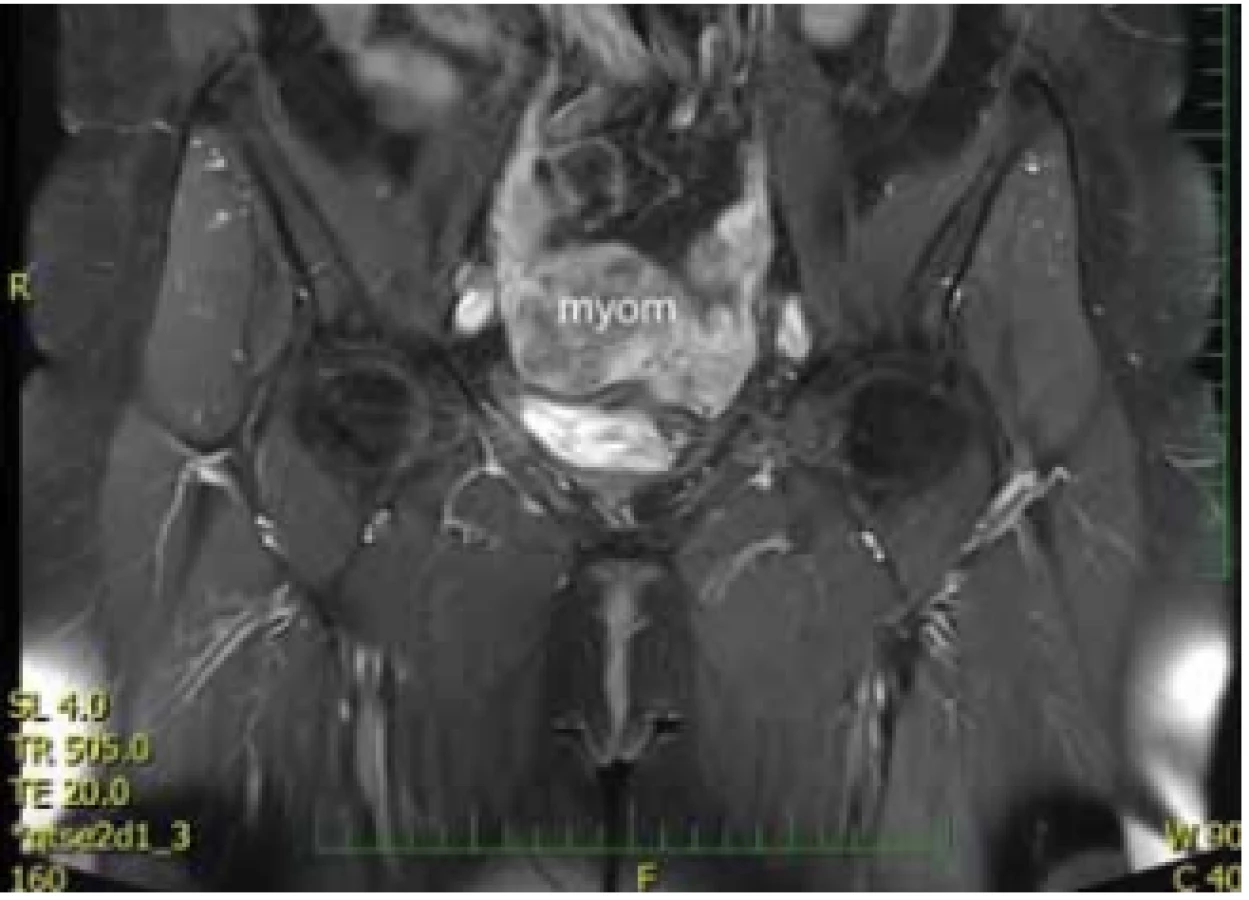
Fig. 4. Finding of magnetic resonance examination – sagittal
view.
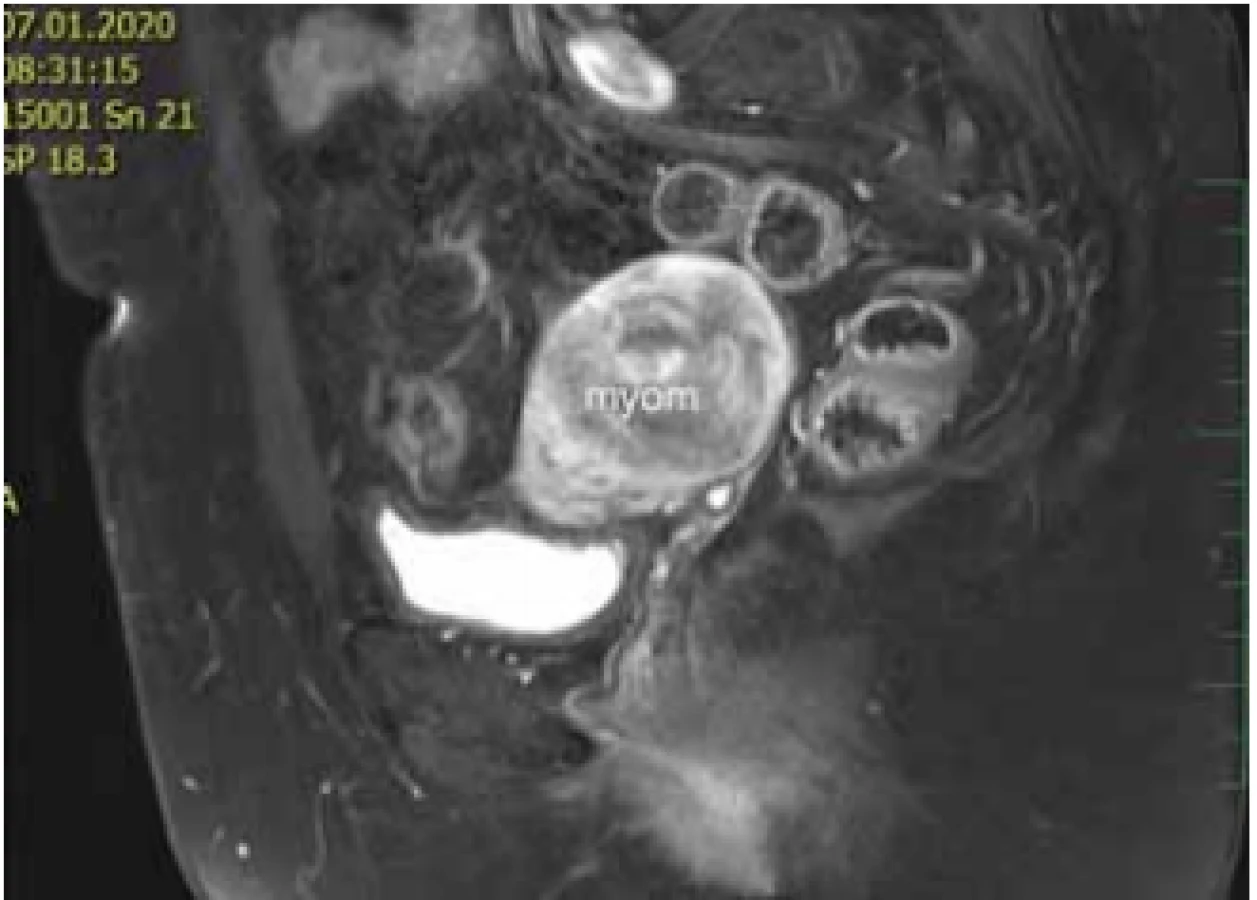
Fig. 5. Perioperative finding of fibroids before laparoscopic surgery
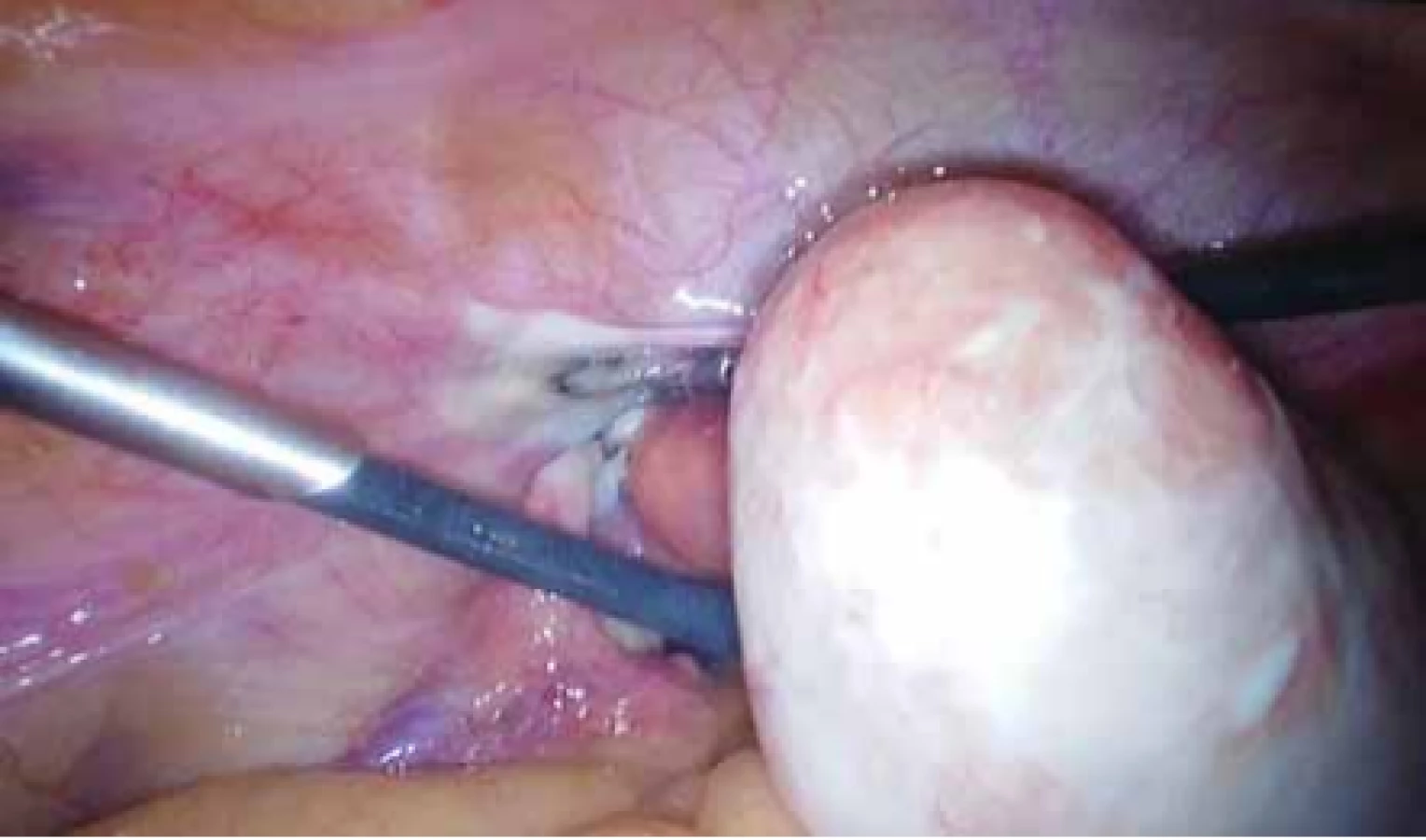
Operace trvala 50 min, proběhla bez komplikací, s krevní ztrátou 50 ml a pacientka byla propuštěna druhý pooperační den. Histopatologické vyšetření popsalo tumory tvořené fascikulárně uspořádanými hladkosvalovými buňkami s hojným hyalinizovaným a fokálně edematózním stromatem odpovídajícím diagnóze leiomyomu.
Diskuze
Syndrom MRKH je vzácné onemocnění znemožňující fyziologickou reprodukci a koitální aktivitu [10,11]. Může však způsobit i akutní nebo chronické gynekologické stavy, které vyžadují chirurgickou intervenci. Ve světové literatuře je popsáno jen několik kazuistik myomů v rudimentárním rohu dělohy [9], v rámci ČR se jedná o první popsaný případ. Prevalence myomů se pohybuje v období perimenopauzy mezi 40 a 50 % [12]. Přesná etiopatogeneze myomů není zcela známá. Jedná se o estrogen senzitivní tumory, jejichž vznik se dává do souvislosti s genetickou predispozicí, hormonálními a různými růstovými faktory. Vzácný výskyt leiomyomu u žen s MRKH syndromem se dává do souvislosti s přítomností buněk hladkého svalstva v proximálním konci Müllerových kanálků, přestože u nich samotná děloha chybí. Spekuluje se také o zvýšené koncentraci nebo citlivosti estrogenových receptorů ve zbytkovém myometriu [9]. Maligní transformace myomů (leiomyosarkom) je vzácná, pohybuje se v rozmezí 0,2–0,3 % [13]. Není nám známo, že by byla popsána v případě leiomyomu u pacientky s MRKH syndromem. U ženy s vrozenou agenezí dělohy a bolestí v podbřišku sice a priori neuvažujeme o původu bolesti spojeném s děložním myomem nebo jeho komplikacemi (torze, nekróza, útlak okolních orgánů, malignizace apod.), je ale nutné o této eventualitě uvažovat. V rámci diferenciální diagnostiky bolestí břicha u žen s MRKH syndromem přicházejí v úvahu především ovariální tumory (fibrom, adenofibrom, endometriom, primární či sekundární solidní ovariální novotvary). Vodítkem k diferenciaci ovariálních tumorů od pendulujících myomů bývá často znázornění cévní spojky s myometriem [12], což může být v námi zmiňovaném případu problematické, a proto hodnotíme především denzitu vyšetřované tkáně a stupeň vaskularizace vyšetřovaného útvaru. Při diagnostice útvarů nejasné etiologie v této lokalizaci bychom neměli zapomínat na gastrointestinální stromální tumor (GIST), který se vyskytuje zejména ve středním věku a vychází nejčastěji ze žaludku, ale existuje i jeho intestinální forma [13]. V úvahu připadají i extravezikálně rostoucí leiomyomy močového měchýře nebo struktury maligních lymfomů. Všechny zmíněné eventuality mohou být asymptomatické (alespoň dočasně) nebo působit potíže. V některých situacích (torze, nekróza, krvácení u maligních prorůstajících tumorů, ileus apod.) mohou způsobit náhlou příhodu břišní.
Závěr
Popsali jsme neobvyklý případ myomatózy, která vycházela z rudimentárních děložních rohů u ženy s MRKH syndromem, z nichž byl jeden leiomyom torkvovaný o 360°. Tyto stavy jsou sice vzácné, ale mohou být příčinou chronických i akutních gynekologických stavů vyžadujících chirurgické řešení.
Obdrženo/Submitted: 17. 11. 2020
Přijato/Accepted: 28. 12. 2020
doc. MUDr. Zlatko Pastor, Ph.D.
Gynekologicko-porodnická klinika
2. LF UK a FN Motol
V Úvalu 84
150 06 Praha 5
Sources
1. Aittomäki K, Eroila H, Kajanoja P. A population-based study of the incidence of Müllerian aplasia in Finland. Fertil Steril 2001; 76 (3): 624–625. doi: 10.1016/s0015-0282 (01) 01 963-x.
2. Valappil S, Chetan U, Wood N et al. Mayer-Rokitansky-Küster-Hauser syndrome: diagnosis and management. Obstet Gynaecol 2012; 14 (2): 93–98. doi: 10.1111/j.1744-4667.2012.00097.x.
3. Hentrich T, Koch A, Weber N et al. The endometrial transcription landscape of MRKH syndrome. Front Cell Dev Biol 2020; 8: 572281. doi: 10.3389/fcell.2020.572281.
4. McQuillan SK, Grover SR. Systematic review of sexual function and satisfaction following the management of vaginal agenesis. Int Urogynecol J 2014; 25 (10): 1313–1320. doi: 10.1007/ s00192-013-2316-3.
5. Chmel R jr, Pastor Z, Mužík M et al. Syndrom Mayer-Rokitansky-Küster-Hauser – ageneze dělohy a pochvy: aktuální znalosti a terapeutické možnosti. Ceska Gynekol 2019; 84 (5): 376–383.
6. Sultan C, Biason-Lauber A, Philibert P. Mayer-Rokitansky-Kuster-Hauser syndrome: recent clinical and genetic findings. Gynecol Endocrinol 2009; 25 (1): 8–11. doi: 10.1080/09513590 802288291.
7. Oppelt PG, Lermann J, Strick R et al. Malformations in a cohort of 284 women with Mayer-Rokitansky-Küster-Hauser syndrome (MRKH). Reprod Biol Endocrinol 2012; 10: 57. doi: 10.1186/1477-7827-10-57.
8. Beecham CT, Skiendzielewski J. Myoma in association with Mayer-Rokitansky-Kuster syndrome. Am J Obstet Gynecol 1977; 129 (3): 346–348. doi: 10.1016/0002-9378 (77) 90801-8.
9. Kundu K, Cohen AW, Goldberg J. Acute torsion of uterine remnant leiomyoma with Mayer-Rokitansky-Küster-Hauser syndrome. Fertil Steril 2014; 102 (2): 607–609. doi: 10.1016/j.fertnstert.2014.04.034.
10. Chmel R, Pastor Z, Matěcha J et al. Uterine transplantaion in the era of successful childbirths from living and deceased donor uteri: current challenges. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2020; 164 (1): 115–120. doi: 10.5507/bp.2019. 020.
11. Pastor Z, Froněk J, Nováčková M et al. Sexual life of women with Mayer-Rokitansky-Küster-Hauser syndrome after laparoscopic Vecchietti vaginoplasty. Sex Med 2017; 5 (2): e106–e113. doi: 10.1016/j.esxm.2016.12.003.
12. Mára M, Frühauf F, Lisá Z. Diferenciální diagnostika děložních myomů. In: Čepický P et al (eds). Kapitoly z diferenciální diagnostiky v gynekologii a porodnictví. Praha: Grada Publishing 2018: 123–128.
13. Stewart EA, Laughlin-Tommaso SK, Catherino WH et al. Uterine fibroids. Nat Rev Dis Primers 2016; 2: 16043. doi: 10.1038/nrdp. 2016.43.
Labels
Paediatric gynaecology Gynaecology and obstetrics Reproduction medicineArticle was published in
Czech Gynaecology

2021 Issue 1
Most read in this issue
- Nežádoucí účinky PARP inhibitorů
- Kombinovaná peripartálna separácia symfýzy a sakroiliakálneho kĺbu
- Aspekty výběru embryí a jejich příprava pro vznik lidských embryonálních kmenových buněk určených k humánní terapii
- Poruchy příjmu potravy v ambulanci gynekologa pro děti a dospívající