
Význam metylace v genomu lidského papillomaviru 16 u karcinomu děložního hrdla
The Significance of Methylation in HPV16 Genome to Cervix Cancerogenesis
Objective:
To summarize the significance of methylation in HPV16 genome to cervix cancerogenesis.
Design:
Review.
Setting:
Department of Oncological and Experimental Pathology, Masaryk Memorial Cancer Institute, Brno.
Subject and Method:
Human papillomaviruses, especially HPV16, are the most frequent causative agents of cervical intraepithelial neoplasia and cervical carcinoma. Their ability to initiate transformation of infected epithelial cells fully depends up production of viral early phase proteins E6 and E7. Affected keratinocytes activate defensive mechanisms based on inhibition of viral DNA transcription by changes in chromatin structure like DNA methylation or histon deacetylation and therefore prevent transcriptional factors from binding to target promoters and from the production of viral oncoproteins.
Conclusion:
Research into epigenetic mechanisms of gene silencing clearly showed their important roles in etiology of cancer. Recent findings confirm the significance of methylation of HPV16 oncogenes leading to block of neoplastic transformation, and simultaneously they indicate new therapeutic possibilities linked with reactivation of methylated tumor supressors.
Key words:
cervix carcinoma, HPV16, oncoproteins E6 and E7, methylation
Autoři:
P. Hublarová; R. Hrstka; B. Vojtěšek
Působiště autorů:
Oddělení onkologické a experimentální patologie, Masarykův onkologický ústav, Brno
Vyšlo v časopise:
Ceska Gynekol 2008; 73(2): 87-92
Souhrn
Cíl studie:
Shrnutí poznatků o významu metylací v genomu HPV16 při kancerogenezi děložního hrdla.
Typ studie:
Souhrnný článek.
Název a sídlo pracoviště:
Oddělení onkologické a experimentální patologie, Masarykův onkologický ústav, Brno.
Předmět a metoda studie:
Lidské papillomaviry v čele s HPV16 jsou nejčastějším původcem cervikálních intraepiteliálních lézí a karcinomu děložního hrdla. Jejich schopnost iniciovat neoplastickou transformaci infikovaných epiteliálních buněk plně závisí na produkci funkčních proteinů rané fáze exprese E6 a E7. Infikované keratinocyty aktivují obranné mechanismy založené především na inhibici transkripce virové DNA pomocí změn ve struktuře chromatinu, jako jsou metylace DNA, deacetylace histonů aj., což zabrání přístupu transkripčních faktorů k promotorům a zablokuje tvorbu virových proteinů.
Závěr:
Výzkum epigenetických mechanismů „umlčování“ genů prokázal jejich důležitou roli v etiologii nádorových onemocnění. Nejnovější poznatky potvrzují význam metylací onkogenů nesených HPV16, které vedou k zablokování neoplastické transformace a současně naznačují nové terapeutické možnosti spojené s reaktivací metylovaných nádorových supresorů.
Klíčová slova:
karcinom děložního hrdla, HPV16, onkoproteiny E6 a E7, metylace
LIDSKÉ PAPILLOMAVIRY
Infekce způsobené lidskými papillomaviry (HPV) vyvolávají benigní i maligní změny epitelu děložního hrdla nádorovou transformací bazálních keratinocytů. Rozlišují se dvě skupiny HPV: i) HPV nízkého rizika, které jsou původci benigních kožních změn typu bradavic aj.; ii) HPV vysokého rizika, které zodpovídají za vznik karcinomů v oblasti genitálu, ORL a dalších [48]. HPV vysokého rizika bývají rovněž identifikovány např. v karcinomech prsu, kolorektálních karcinomech, ale jejich podíl na nádorové transformaci není doposud jednoznačně prokázán [12, 55].
Nádory děložního hrdla způsobuje přibližně 15 typů HPV vysokého rizika [62], které jsou zodpovědné za 99 % těchto nádorů [75]. HPV 16 je nejčastější a vyskytuje se přibližně u 50 % nádorů [62]. Příčinou je pravděpodobně schopnost delší perzistence HPV16 v buňce [21, 32, 33], což je stav nezbytný pro vznik prekancerózních lézí na děložním hrdle [62]. Samotná infekce vysoce rizikovými HPV obvykle nestačí ke vzniku karcinomů [48]. Na kancerogenezi děložního hrdla se podílí množství dalších kofaktorů – např. užívání hormonální antikoncepce, promiskuita, kouření aj. [48]. Incidence v České republice je 18,7 případů karcinomu děložního hrdla na 100 000 žen ročně [8].
HPV infikují keratinocyty bazální vrstvy epitelu, přičemž využívají buněčný aparát k vlastní replikaci [17]. Replikace probíhá výhradně v jádře [18] v synchronizaci s replikací buněčné DNA. Během infekce je virová DNA ve formě epizomů v přibližně 50 kopiích na buňku [67]. Exprese virových genů včetně samotné replikace je regulována diferenciací keratinocytů zatím neznámým mechanismem [80].
BIOLOGIE HPV
HPV jsou malé (přibližně 55 nm) neobalené dsDNA viry s vysokou afinitou pro lidské epitely a sliznice [80]. Genom HPV je tvořen cirkulární dsDNA o délce okolo 8 kbp [48] a obsahuje 2 kódující oblasti a regulační oblast URR (Upstream Regulatory Region) nebo-li LCR (Long Control Region) [80].
Virový genom obsahuje dva typy genů: i) geny kódující regulační proteiny – jsou aktivní v rané fázi exprese v nediferencovaném keratinocytu (E1, E2, E4, E5, E6 a E7); ii) geny kódující strukturní proteiny, které se přepisují v pozdní fázi exprese v diferencovaném keratinocytu (L1 a L2). Proteiny E1 a E2 regulují replikaci virové DNA a expresi proteinů rané fáze. Protein E4 se uplatňuje při produktivní infekci a způsobuje zhroucení cytokeratinových filament. Zbylé proteiny E5, E6 a E7 jsou prokázané onkogeny a zodpovídají za imortalizaci a transformaci buněk [80].
Onkogeny E6 a E7 leží ve stejném otevřeném čtecím rámci, který je přepisován z raného promotoru P97 [17]. Tento promotor leží na konci LCR a je pod kontrolou buněčného transkripčního faktoru SP1 a virového regulátoru E2 [15, 69]. Jeho aktivita je stimulována tzv. „enhancerem“ (zesilovačem) s vazebnými místy pro další transkripční faktory [2]. Oba proteiny vznikají sestřihem jedné polycistronní mRNA a následnou translací sestřižených částí [64].
KANCEROGENEZE DĚLOŽNÍHO HRDLA – PROTEINY RANÉ FÁZE EXPRESE E6 A E7
Pro replikaci viru jsou kriticky důležité proteiny E6 a E7, které v buňce interagují s různými buněčnými proteiny [48]. Bývají trvale exprimovány v HPV pozitivních nádorech děložního hrdla [47]. Transkripce E6 a E7 je závislá na mnoha transkripčních faktorech a epigenetických mechanismech [27]. Progrese nádoru může vzejít ze stimulace exprese onkoproteinů steroidy [26, 35], z delece transkripčních „silencerů“ [44] či integrací HPV genomu do buněčné DNA [3, 63, 68], kdy se transkripční stimulátor „nuclear matrix attachment region“ dostane do blízkosti P97 a razantně zvýší jeho aktivitu [68].
Úkolem E6 je vyřadit z funkce protein p53, podobně protein E7 inaktivuje protein pRb [80]. Protein p53 funguje jako tzv. „strážce genomu“, který se v buňkách běžně nachází ve velmi nízkých hladinách. Při poškození buňky či při stresu v G1-fázi buněčného cyklu dochází k fosforylaci a stabilizaci p53 a takto aktivovaný p53 následně zablokuje buněčný cyklus pomocí proteinu p21Waf1 nebo přímo indukuje apoptózu.
Protein pRb je uzlovým regulátorem přechodu buněk do S-fáze. V aktivním hypofosforylovaném stavu vytváří pRb komplex s transkripčním faktorem E2F, čímž blokuje funkci E2F v iniciaci transkripce genů nutných pro vstup buněk do S-fáze. Při funkčním vyřazení p53 a pRb vstupuje buňka do S-fáze nekontrolovaně a zahajuje replikaci převážně virové DNA [19] (obr. 1).
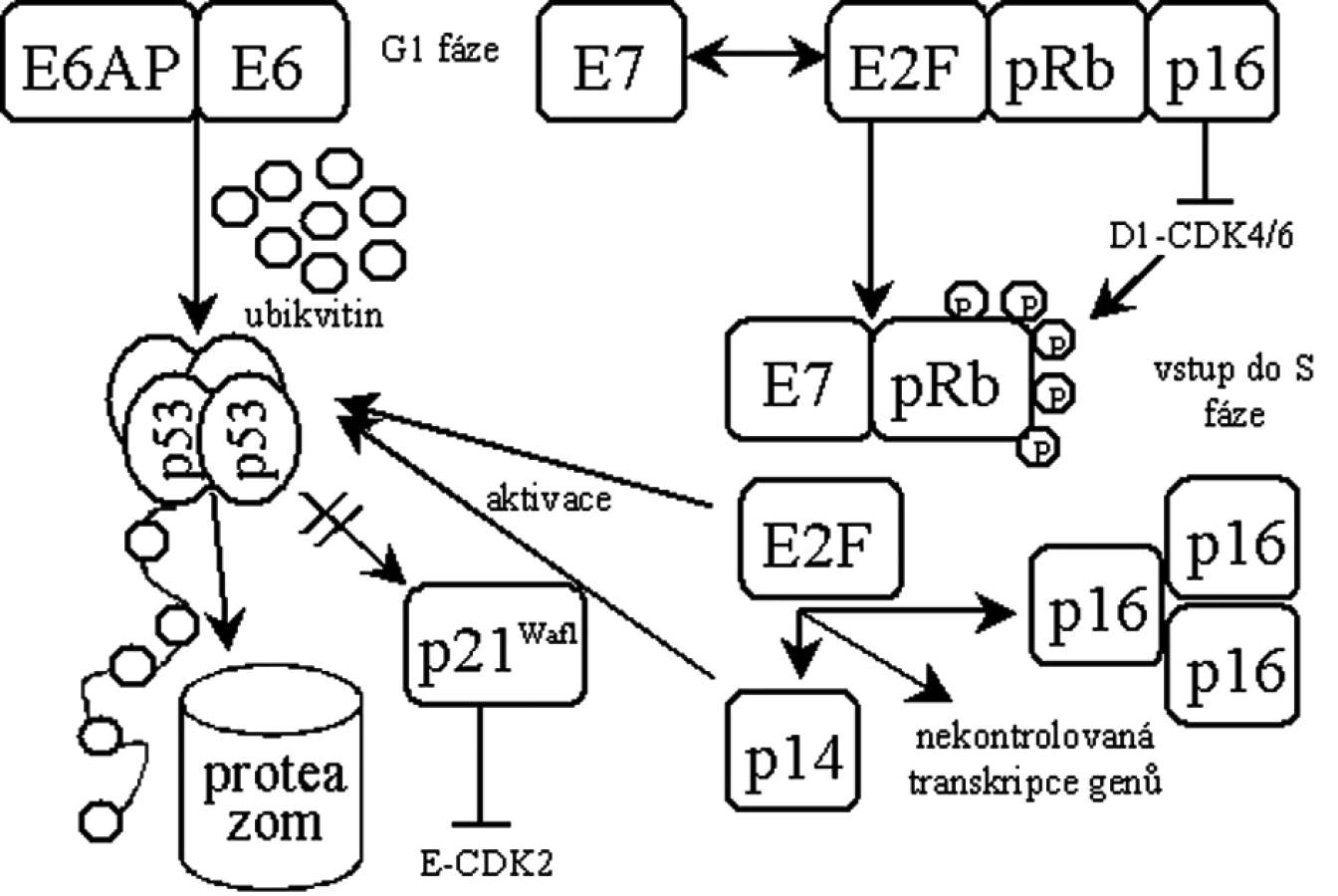
Protein E6 se skládá ze 158 aminokyselin tvořících dvě domény se zinkovými prsty [47]. Kromě schopnosti tvořit komplex s p53 vykazuje antiapoptotické vlastnosti [47, 76]. Pro odbourání p53 se E6 spojuje s buněčnou ubikvitin ligázou E6AP [34] a v komplexu specificky ubikvitinují p53 [61], což navodí jeho zrychlenou degradaci v proteazomech [47]. Byl však popsán i mechanismus odbourávání p53 nezávisle na ubikvitinu [9]. Dalším kritickým krokem při transformaci buňky je pak aktivace buněčné telomerázy proteinem E6 [18, 29, 42].
Protein E7 je primárně jaderný protein o velikosti 98 aminokyselin. Ke své aktivitě potřebuje dimerizaci [46], jinak je velice rychle odbouráván [58]. E7 obsahuje konzervované domény shodné s doménami proteinu E1A u adenovirů a Tag u SV40, které jsou obecně zodpovědné za transformační aktivitu a vazbu s důležitými regulačními proteiny buňky. Nejdůležitější aktivitou E7 je asociace s rodinou retinoblastomových (Rb) tumor supresorů [47]. E7 tvoří přednostně komplexy s hypofosforylovaným pRb, což vede k inaktivaci pRb [10] a k jeho odbourání v proteazomech [4]. Současně je vazbou E7 na pRb uvolněn transkripční faktor E2F [45]. Tento stav by za normálních podmínek vedl k p53 indukované apoptóze, ale u buněk infikovaných HPV je p53 degradován E6 a buňka pokračuje v dělení [70], důsledkem je pak přerušení diferenciace keratinocytu [16].
Dysfunkce pRb vyvolaná E7 mimo jiné způsobuje i nadměrnou expresi inhibitoru cyklin-dependentních kináz p16ink4a [39, 54], který se váže k pRb, a ve zdravých buňkách tak zabraňuje fosforylaci pRb, čímž reguluje buněčný cyklus [6, 43, 60]. Ke zvýšené expresi p16ink4a může docházet navázáním E7 na pRb za současného uvolnění E2F, který iniciuje transkripci p16ink4a [25] nebo pouze samotnou vazbou E7 na pRb [41]. Vzhledem k jeho významu se o p16ink4a uvažuje jako o potenciálním biomarkeru infekce vysoce rizikovými HPV spojené se vznikem CIN 2-3 [28, 49, 50, 74]. Nicméně, vzácně existují i nádory, ve kterých není p16ink4a nadměrně exprimován [73].
E7 má schopnost vyřadit z funkce inhibitory cyklin-dependentních kináz - proteiny p21Waf1 [24, 36] a p27 [77], což se současnou inaktivací pRb zabrání zablokování buněčného cyklu a neumožní diferenciaci keratinocytu.
EPIGENETICKÉ MECHANISMY „UMLČOVÁNÍ“ GENŮ
Základní jednotkou chromatinových struktur je nukleozom – dimer složený ze dvou identických komplexů histonů H2A, H2B, H3 a H4 [56]. Histony H3 a H4 jsou vysoce nabité a zodpovědné za vazbu k DNA [11]. Struktura chromatinu je regulována různými modifikacemi histonů, jako je acetylace, metylace, fosforylace, ubikvitinace či sumoylace [66, 79]. Právě modifikace určují, zda bude DNA přístupná pro transkripci, reparaci, DNA replikaci či segregaci chromozomů [11]. Acetylace přímo ovlivňuje přístupnost chromatinu pro transkripční faktory. Histonacetyltransferázy (HAT) zajišťují přenos acetylu na histony a otevření struktury chromatinu, naopak histondeacetylázy (HDAC) jej odbourávají [53], což se děje v součinnosti s DNA metylací (obr. 2).
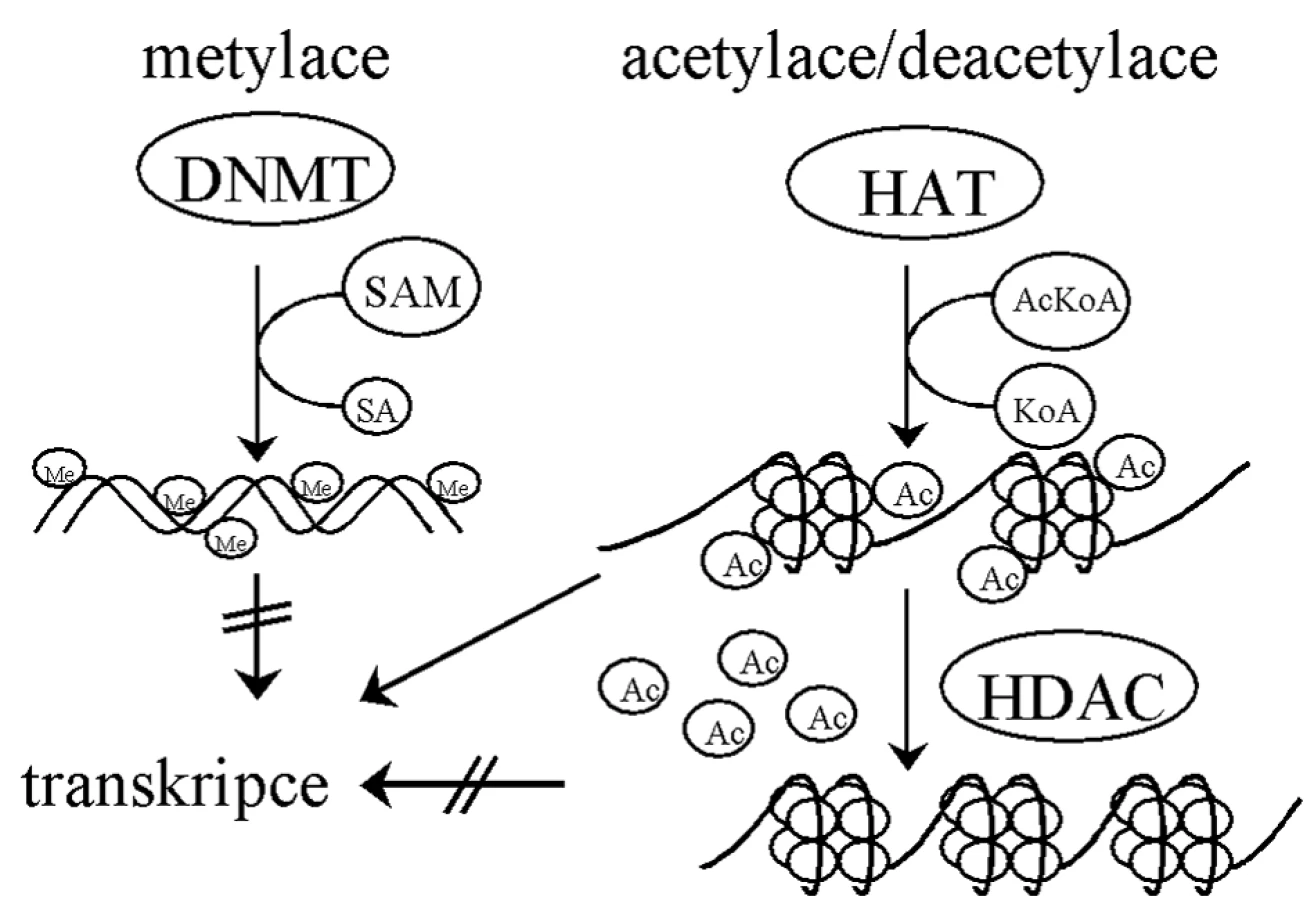
Mechanismus metylace DNA spočívá v navázání metylových skupin S-adenosylmethioninu pomocí metyltransferáz (DNMTs) na uhlík č. 5 pyridiminového kruhu cytosinu v CpG dinukleotidech [11]. Metylace DNA jsou katalyzovány DNMT3a a DNMT3b [52] a objevují se během vývoje de novo. Po každém buněčném dělení je DNA dceřiné buňky semimetylovaná. DNMT1 udržuje stav metylace nově vytvořené molekuly DNA podle vzoru rodičovské molekuly [22, 57].
Metylace DNA jsou obecně prvním signálem inaktivace genů [51] a uplatňují se hlavně v promotorových a regulačních oblastech, kde brání vazbě transkripčních faktorů [37]. Rovněž samotná chromatinová struktura je schopna určovat DNA metylaci, neboť proteiny účastnící se „umlčování“ chromatinu jsou schopné přitahovat metyltransferázy ke kódujícím sekvencím [5, 23, 72].
Během transformace buňky je aktivita DNMT1 zvyšována přímou vazbou proteinu E7 přes jeho transformační doménu. Zvýšená aktivita DNMT1 způsobuje neregulovanou metylaci genomu provázenou buněčnou transformací, což vede ke stejným následkům jako umlčení tumor supresorových genů [7].
METYLACE V GENOMU HPV16
U HPV16 jsou popsány tři druhy metylace, které se mohou vyskytovat v buňkách současně: i) metylace epizomálních forem virové DNA v buňkách asymptomatických stěrů, ii) metylace tandemově začleněných genomů a iii) de novo metylace především ve slizničním epitelu somatických tkání [2, 40, 59, 71]. Obecně neexistují místa, která by byla metylována vždy nebo naopak nikdy. Metylace se vyskytují nejčastěji v konzervovaných oblastech DNA tvořených nejčastěji 3 až 8, ale někdy i více sousedními CpG. Naopak izolované CpG jsou metylovány jen zřídka [38].
Metylace se rovněž vyskytují podle organizace DNA a histonů. Nukleozomálně organizované segmenty DNA mají spíše tendenci k metylaci na rozdíl od nukleozomálních spojů. U HPV16 se metylace CpG nejčastěji vyskytují v LCR oblasti a v části L1 otevřeného čtecího rámce [1, 2].
Metylace jsou identifikovány u všech patologických stavů děložního hrdla, ovšem s rozdílnou frekvencí. Nízké frekvence metylací u CIN, kde se HPV normálně replikuje epizomálně, mají pravděpodobně původ v transkripčně aktivních buněčných populacích, u kterých se iniciuje neoplastický proces a narůstá u nich počet kopií genomu viru [65]. Přítomnost metylací genomu HPV u karcinomů je běžnější než u lézí nižšího stupně, což zřejmě souvisí s celkovými změnami v metylačních vzorcích nádorové buňky [20, 78]. U vysokého procenta karcinomů se genomy HPV16 vyskytují v integrovaném stavu [13, 63], často ještě v tandemových repeticích, avšak jsou obvykle z velké části metylované a jen několik málo genů zůstává transkripčně aktivních [2, 71].
Klinická studie zaměřená na stanovení četnosti metylací LCR a genu E6 podle patologického typu uvádí frekvence 52 % u asymptomatických stěrů, 21,7 % u prekancerózních stadií a 6,1 % u invazivních karcinomů. Současně bylo zjištěno, že studovaná oblast zesilovače transkripce a promotoru obsahuje 11 CpG ostrůvků. Bisulfitovou metodou byl určen stav metylace, přičemž u asymptomatických stěrů byly CpG metylované, zatímco u cervikálních lézí zůstávaly všechny nebo alespoň část CpG nemetylovaných [2]. Zdá se, že metylace CpG jsou zahrnuty nejen v biologii HPV16, ale i v etiologii karcinomu děložního hrdla. Metylace může představovat i skrytou strategii HPV, jak zůstat dlouhodobě v subklinické fázi. Tyto výsledky podporují možnost využít stanovení metylace v oblasti zesilovače transkripce a promotoru jako relevantní marker vývoje karcinomu děložního hrdla [2].
Naše vlastní výsledky potvrzují hypotézu zablokování neoplastické transformace metylací CpG ostrůvků, zatímco demetylace se projeví jako příčina či následek nádorové progrese [2]. Tato komplexní analýza byla provedena v souboru tvořeném zdravými ženami a pacientkami s neoplastickými změnami děložního hrdla. Stanovením metylace bychom rádi doplnili cytologické vyšetření přítomnosti HPV při diagnostice děložního hrdla a přispěli tak k jejímu zpřesnění. Jelikož metoda monitoruje aktivitu virového genomu, je možné pravidelným sledováním průběžně stanovovat riziko vzniku nádoru pro jednotlivé pacientky.
EPIGENETICKÉ TERAPIE
Podstatou těchto terapií je aktivace transkripčně umlčených tumor supresorových genů, které by byly schopné navodit apoptózu nádorové buňky. Používají se inhibitory DNMT (např. 5-azacytidin, hydrolazin) či HDAC (např. vorinostat, valproát), schopné navodit demetylaci a reaktivaci reprimovaných tumor supresorových genů [31].
Avšak velké nebezpečí představuje plošné použití inhibitorů metylace [31], neboť v infikovaných tkáních může dojít k reaktivaci genů kódujících virové transformační proteiny, čímž může docházet k vyvolání nádorového onemocnění či jeho rychlejší progresi nejen u karcinomu děložního hrdla ale i jiných nádorů s virovou etiologií. Na druhou stranu poslední molekulárně-farmakologická studie zkoumající možnosti léčby pomocí hydrazinu a valproátu včetně negativních účinků prokázala, že tato terapie nezpůsobuje u pacientek s karcinomem cervixu zvýšení exprese E6 a E7, ta zůstává stejná, ale naopak dochází k indukci exprese p53 [14].
Tato práce byla podporována granty MZ0MOU2005 a MŠMT LC06035.
RNDr. Bořivoj Vojtěšek, DrSc.
Oddělení onkologické a experimentální patologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
Zdroje
1. Badal, S., Badal, V., Calleja-Macias, IE., et al. The human papillomavirus-18 genome is efficiently targeted by cellular DNA methylation. Virology, 2004, 324, 2. p. 483-492.
2. Badal, V., Chuang, LS., Tan, EH., et al. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: genomic hypomethylation correlates with carcinogenic progression. J Virol, 2003, 77, 11, p. 6227-6234.
3. Baker, CC., Phelps, WC., Lindgren, V., et al. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J Virol, 1987, 61, 4, p. 962-971.
4. Boyer, SN., Wazer, DE., Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res, 1996. 56, 20, p. 4620-4.
5. Brenner, C., Deplus, R., Didelot, C., et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. Embo J, 2005, 24, 2, p. 336-346.
6. Bringold, F., Serrano, M. Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol, 2000. 35, 3, p. 317-329.
7. Burgers, WA., Blanchon, L., Pradhan, S., et al. Viral oncoproteins target the DNA methyltransferases. Oncogene, 2007. 26, 11, p. 1650-1655.
8. Cabrnochová, http://www.vakciny.net/doporucene_ockovani/ HPV.html#1, Brno, leden 2007 On-line 17. 12. 2007.
9. Camus, S., Menendez, S., Cheok, CF., et al. Ubiquitin-independent degradation of p53 mediated by high-risk human papillomavirus protein E6. Oncogene, 2007, 26, 28, p. 4059-4070.
10. Cobrinik, D., Dowdy, SF., Hinds, PW., et al. The retinoblastoma protein and the regulation of cell cycling. Trends Biochem Sci, 1992, 17, 8, p. 312-315.
11. D’Alessio, AC., Szyf, M. Epigenetic tete-a-tete: the bilateral relationship between chromatin modifications and DNA methylation. Biochem Cell Biol, 2006, 84, 4, p. 463-476.
12. Damin, AP., Karam, R., Zettler, CG., et al. Evidence for an association of human papillomavirus and breast carcinomas. Breast Cancer Res Treat, 2004, 84, 2, p. 131-137.
13. Daniel, B., Mukherjee, G., Seshadri, L., et al. Changes in the physical state and expression of human papillomavirus type 16 in the progression of cervical intraepithelial neoplasia lesions analysed by PCR. J Gen Virol, 1995, 76, Pt 10, p. 2589-2593.
14. de la Cruz-Hernandez, E., Perez-Cardenas, E., Contreras-Paredes, A., et al. The effects of DNA methylation and histone deacetylase inhibitors on human papillomavirus early gene expression in cervical cancer, an in vitro and clinical study. Virol J, 2007, 4, p. 18.
15. Demeret, C., Desaintes, C., Yaniv, M., et al. Different mechanisms contribute to the E2-mediated transcriptional repression of human papillomavirus type 18 viral oncogenes. J Virol, 1997, 71, 12, p. 9343-9349.
16. Doorbar, J. The papillomavirus life cycle. J Clin Virol, 2005, 32, Suppl 1, p. S7-S15.
17. Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond), 2006, 110, 5, p. 525-541.
18. Elenbaas, B., Spirio, L., Koerner, F., et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev, 2001, 15, 1, p. 50-65.
19. Fehrmann, F., Laimins, LA. Human papillomaviruses: targeting differentiating epithelial cells for malignant transformation. Oncogene, 2003. 22, 33, p. 5201-5207.
20. Feng, Q., Balasubramanian, A., Hawes, SE., et al. Detection of hypermethylated genes in women with and without cervical neoplasia. J Natl Cancer Inst, 2005, 97, 4, p. 273-282.
21. Franco, EL., Villa, LL., Sobrinho, JP., et al. Epidemiology of acquisition and clearance of cervical human papillomavirus infection in women from a high-risk area for cervical cancer. J Infect Dis, 1999, 180, 5, p. 1415-1423.
22. Fuks, F. DNA methylation and histone modifications: teaming up to silence genes. Curr Opin Genet Dev, 2005, 15, 5, p. 490-495.
23. Fuks, F., Burgers, WA., Brehm, A., et al. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet, 2000, 24, 1, p. 88-91.
24. Funk, JO., Waga, S., Harry, JB., et al. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev, 1997, 11, 16, p. 2090-2100.
25. Giarre, M., Caldeira, S., Malanchi, I., et al. Induction of pRb degradation by the human papillomavirus type 16 E7 protein is essential to efficiently overcome p16INK4a-imposed G1 cell cycle Arrest. J Virol, 2001, 75, 10, p. 4705-4712.
26. Gloss, B., Bernard, HU., Seedorf, K., et al. The upstream regulatory region of the human papilloma virus-16 contains an E2 protein-independent enhancer which is specific for cervical carcinoma cells and regulated by glucocorticoid hormones. Embo J, 1987, 6, 12, p. 3735-3743.
27. Gloss, B., Chong, T., Bernard, HU. Numerous nuclear proteins bind the long control region of human papillomavirus type 16: a subset of 6 of 23 DNase I-protected segments coincides with the location of the cell-type-specific enhancer. J Virol, 1989, 63, 3, p. 1142-1152.
28. Guo, M., Hu, L., Baliga, M., et al. The predictive value of p16(INK4a) and hybrid capture 2 human papillomavirus testing for high-grade cervical intraepithelial neoplasia. Am J Clin Pathol, 2004, 122, 6, p. 894-901.
29. Hahn, WC., Counter, CM., Lundberg, AS., et al. Creation of human tumour cells with defined genetic elements. Nature, 1999, 400, 6743, p. 464-468.
30. Helt, AM., Funk, JO., Galloway, DA. Inactivation of both the retinoblastoma tumor suppressor and p21 by the human papillomavirus type 16 E7 oncoprotein is necessary to inhibit cell cycle arrest in human epithelial cells. J Virol, 2002, 76, 20, p. 10559-10568.
31. Herman, JG., Baylin, SB. Promoter-region hypermethylation and gene silencing in human cancer. Curr Top Microbiol Immunol, 2000, 249, p. 35-54.
32. Hildesheim, A., Schiffman, MH., Gravitt, PE., et al. Persistence of type-specific human papillomavirus infection among cytologically normal women. J Infect Dis, 1994, 169, 2, p. 235-240.
33. Ho, GY., Burk, RD., Klein, S., et al. Persistent genital human papillomavirus infection as a risk factor for persistent cervical dysplasia. J Natl Cancer Inst, 1995, 87, 18, p. 1365-1371.
34. Huibregtse, JM., Scheffner, M., Howley, PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. Embo J, 1991, 10, 13, p. 4129-4135.
35. Chan, WK., Klock, G., Bernard, HU. Progesterone and glucocorticoid response elements occur in the long control regions of several human papillomaviruses involved in anogenital neoplasia. J Virol, 1989. 63, 8, p. 3261-3269.
36. Jones, DL., Alani, RM., Munger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev, 1997, 11, 16, p. 2101-2111.
37. Jones, PL., Wade, PA., Wolffe, AP. Purification of the MeCP2/Histone Deacetylase Complex from Xenopus laevis. Methods Mol Biol, 2001, 181, p. 297-307.
38. Kalantari, M., Calleja-Macias, IE., Tewari, D., et al. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J Virol, 2004, 78, 23, p. 12762-12772.
39. Khleif, SN., DeGregori, J., Yee, CL., et al. Inhibition of cyclin D-CDK4/CDK6 activity is associated with an E2F-mediated induction of cyclin kinase inhibitor activity. Proc Natl Acad Sci U S A, 1996, 93, 9, p. 4350-4351.
40. Kim, K., Garner-Hamrick, PA., Fisher, C., et al. Methylation patterns of papillomavirus DNA, its influence on E2 function, and implications in viral infection. J Virol, 2003, 77, 23, p. 12450-12459.
41. Kim, YT., Zhao, M. Aberrant cell cycle regulation in cervical carcinoma. Yonsei Med J, 2005, 46, 5, p. 597-613.
42. Klingelhutz, AJ., Foster, SA., McDougall, JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature, 1996, 380, 6569, p. 79-82.
43. Li, Y., Nichols, MA., Shay, JW., et al. Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb. Cancer Res, 1994, 54, 23, p. 6078-6082.
44. May, M., Dong, XP., Beyer-Finkler, E., et al. The E6/E7 promoter of extrachromosomal HPV16 DNA in cervical cancers escapes from cellular repression by mutation of target sequences for YY1. Embo J, 1994, 13, 6, p. 1460-1466.
45. Munger, K., Baldwin, A., Edwards, KM., et al. Mechanisms of human papillomavirus-induced oncogenesis. J Virol, 2004, 78, 21, p. 11451-11460.
46. Munger, K., Basile, JR., Duensing, S., et al. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene, 2001. 20, 54, p. 7888-7898.
47. Munger, K., Howley, PM. Human papillomavirus immortalization and transformation functions. Virus Res, 2002, 89, 2, p. 213-228.
48. Munoz, N., Castellsague, X., de Gonzalez, AB., et al. Chapter 1: HPV in the etiology of human cancer. Vaccine, 2006, 24S3, p. S1-S10.
49. Murphy, N., Heffron, CC., King, B., et al. p16INK4A positivity in benign, premalignant and malignant cervical glandular lesions: a potential diagnostic problem. Virchows Arch, 2004, 445, 6, p. 610-615.
50. Murphy, N., Ring, M., Killalea, AG., et al. p16INK4A as a marker for cervical dyskaryosis: CIN and cGIN in cervical biopsies and ThinPrep smears. J Clin Pathol, 2003, 56, 1, p. 56-63.
51. Nan, X., Cross, S., Bird, A. Gene silencing by methyl-CpG-binding proteins. Novartis Found Symp, 1998, 214, p. 6-16; discussion 16-21, 46-50.
52. Okano, M., Bell, DW., Haber, DA., et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 1999, 99, 3, p. 247-257.
53. Pan, LN., Lu, J., Huang, BQ. HDAC Inhibitors: A potential new category of anti-tumor agents. Cell Mol Immunol, 2007. 4, 5, p. 337-43.
54. Parry, D., Bates, S., Mann, DJ., et al. Lack of cyclin D-Cdk complexes in Rb-negative cells correlates with high levels of p16INK4/MTS1 tumour suppressor gene product. Embo J, 1995, 14, 3, p. 503-511.
55. Perez, LO., Abba, MC., Laguens, RM., et al. Analysis of adenocarcinoma of the colon and rectum: detection of human papillomavirus (HPV) DNA by polymerase chain reaction. Colorectal Dis, 2005, 7, 5, p. 492-495.
56. Pruss, D., Hayes, JJ., Wolffe, AP. Nucleosomal anatomy—where are the histones? Bioessays, 1995, 17, 2, p. 161-170.
57. Razin, A., Szyf, M. DNA methylation patterns. Formation and function. Biochim Biophys Acta, 1984, 782, 4, p. 331-342.
58. Reinstein, E., Scheffner, M., Oren, M., et al. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: targeting via ubiquitination of the N-terminal residue. Oncogene, 2000, 19, 51, p. 5944-5950.
59. Rosl, F., Arab, A., Klevenz, B., et al. The effect of DNA methylation on gene regulation of human papillomaviruses. J Gen Virol, 1993, 74, Pt 5, p. 791-801.
60. Serrano, M., Hannon, GJ., Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature, 1993, 366, 6456, p. 704-707.
61. Scheffner, M., Werness, BA., Huibregtse, JM., et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell, 1990, 63, 6, p. 1129-1136.
62. Schiffman, M., Castle, PE., Human papillomavirus: epidemiology and public health. Arch Pathol Lab Med, 2003, 127, 8, p. 930-934.
63. Schwarz, E., Freese, UK., Gissmann, L., et al. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature, 1985. 314, 6006, p. 111-114.
64. Stacey, SN., Jordan, D., Williamson, AJ., et al. Leaky scanning is the predominant mechanism for translation of human papillomavirus type 16 E7 oncoprotein from E6/E7 bicistronic mRNA. J Virol, 2000, 74, 16, p. 7284-7297.
65. Stoler, MH., Rhodes, CR., Whitbeck, A., et al. Human papillomavirus type 16 and 18 gene expression in cervical neoplasias. Hum Pathol, 1992, 23, 2, p. 117-128.
66. Strahl, BD., Allis, CD. The language of covalent histone modifications. Nature, 2000, 403, 6765, p. 41-45.
67. Stubenrauch, F., Laimins, LA. Human papillomavirus life cycle: active and latent phases. Semin Cancer Biol, 1999, 9, 6, p. 379-386.
68. Stunkel, W., Huang, Z., Tan, SH., et al. Nuclear matrix attachment regions of human papillomavirus type 16 repress or activate the E6 promoter, depending on the physical state of the viral DNA. J Virol, 2000, 74, 6, p. 2489-2501.
69. Tan, SH., Leong, LE., Walker, PA., et al. The human papillomavirus type 16 E2 transcription factor binds with low cooperativity to two flanking sites and represses the E6 promoter through displacement of Sp1 and TFIID. J Virol, 1994, 68, 10, p. 6411-6420.
70. Thomas, M., Pim, D., Banks, L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene, 1999, 18, 53, p. 7690-7700.
71. Van Tine, BA., Knops, J., Broker, TR., et al. In situ analysis of the transcriptional activity of integrated viral DNA using tyramide-FISH. Dev Biol (Basel), 2001, 106, p. 381-385.
72. Vire, E., Brenner, C., Deplus, R., et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature, 2006, 439, 7078, p. 871-874.
73. Volgareva, G., Zavalishina, L., Andreeva, Y., et al. Protein p16 as a marker of dysplastic and neoplastic alterations in cervical epithelial cells. BMC Cancer, 2004, 4, p. 58.
74. von Knebel Doeberitz, M. New molecular tools for efficient screening of cervical cancer. Dis Markers, 2001, 17, 3, p. 123-128.
75. Walboomers, JM., Jacobs, MV., Manos, MM., et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol, 1999, 189, 1, p. 12-19.
76. Werness, BA., Levine, AJ., Howley, PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science, 1990. 248, 4951, p. 76-79.
77. Zerfass-Thome, K., Zwerschke, W., Mannhardt, B., et al. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene, 1996, 13, 11, p. 2323-2330.
78. Zhang, J., Martins, CR., Fansler, ZB., et al. DNA methylation in anal intraepithelial lesions and anal squamous cell carcinoma. Clin Cancer Res, 2005, 11, 18, p. 6544-6549.
79. Zhang, Y., Reinberg, D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev, 2001, 15, 18, p. 2343-2360.
80. Zheng, ZM., Baker, CC. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front Biosci, 2006, 11, p. 2286-2302.
Štítky
Dětská gynekologie Gynekologie a porodnictví Reprodukční medicínaČlánek vyšel v časopise
Česká gynekologie

2008 Číslo 2
Nejčtenější v tomto čísle
- Indukce potratů ve II. trimestru na Gynekologicko porodnické klinice FN na Bulovce
- Perzistující trofoblastická nemoc v Centru pro trofoblastickou nemoc v ČR v letech 1955 – 2007
- Význam sonografie a hysteroskopie u suspektních nálezů na endometriu menopauzálních žen
- Adrenokortikálne choroby v gravidite