
Cystická adenomatoidná malformácia plodu
Cystic adenomatoid malformation of fetus
Objective:
Presentation of prenataly diagnosed fetus with congenital cystic adenomatoid malformation (CCAM). Summary of clinical and histological findings in fetuses with CCAM, in utero ultrasound diagnosis, prognosis, in utero dispensarisation, timing of delivery and postanatal management.
Design:
Case report.
Settings:
Nemocnica s poliklinikou, Zvolen, a.s., gynekologicko-pôrodnícke oddelenie.
Case report:
In this article we would like to introduce the case report of fetus suffering from CCAM.
Conclusions:
Congenital cystic adenomatoid malformation is a rare congenital disorder. The clinical and histological findings can vary. The diagnose can be made in prenatal period due to the availability of prenatal ultrasound examination.
Keywords:
congenital cystic adenomatoid malformation of lung, prenatal ultrasound diagnosis, polyhydramnion
Authors:
K. Bihariová 1; D. Kobidová 1; T. Bielik 2; P. Molitoris 1
Authors‘ workplace:
Gynekologicko-pôrodnícke oddelenie, NsP Zvolen, a. s., primár MUDr. P. Molitoris
1; Gynekologicko-pôrodnícka klinika, Fakultná nemocnica s poliklinikou F. D. Roosevelta, Banská Bystrica, prednosta doc. MUDr. T. Bielik, PhD.
2
Published in:
Ceska Gynekol 2017; 82(6): 474-477
Overview
Cieľ štúdie:
Prezentácia kazuistiky prenatálne diagnostikovanej cystickej adenomatoidnej malformácie plodu (congenital cystic adenomatoid malformation – CCAM). Zhrnutie klinických a histologických obrazov cystickej adenomatoidnej malformácie u plodov; ultrazvuková diagnostika, prognóza, in utero dispenzarizácia, načasovanie pôrodu a postnatálny management v neonatálnej perióde.
Typ štúdie:
Kazuistika.
Názov a sídlo pracoviska:
Nemocnica s poliklinikou, Zvolen, a.s., gynekologicko-pôrodnícke oddelenie.
Vlastné pozorovanie:
Prípad prenatálne diagnostikovanej cystickej adenomatoidnej malformácie plodu.
Záver:
Kongenitálna cystická adenomatoidná malformácia je vzácna kongenitálna choroba. Môže byť vyjadrená rôznymi klinickými a histologickými obrazmi. Pri dnešnej dostupnosti ultrasonografického skríningu je diagnostika týchto vrodených chýb obvykle posunutá do prenatálneho obdobia.
Klíčová slova:
kongenitálna cystická adenomatoidná malformácia pľúc, prenatálna ultrasonografická diagnostika, polyhydramnion
ÚVOD
CCAM je pomerne vzácna vrodená vývojová chyba. Prvýkrát ju popísali autori Ch´in a Tang v roku 1949. Vyskytuje sa s incidenciou 1 : 10 000 až 1 : 35 000 živonarodených detí [7, 9, 12], ale je pravdepodobné, že incidencia je o niečo vyššia, vzhľadom na možnú spontánnu intrauterinnú regresiu nálezu a pre možnosť rozhodnúť sa pre abort z genetickej indikácie. Prezentujeme prípad plodu so závažným CCAM.
VLASTNÉ POZOROVANIE
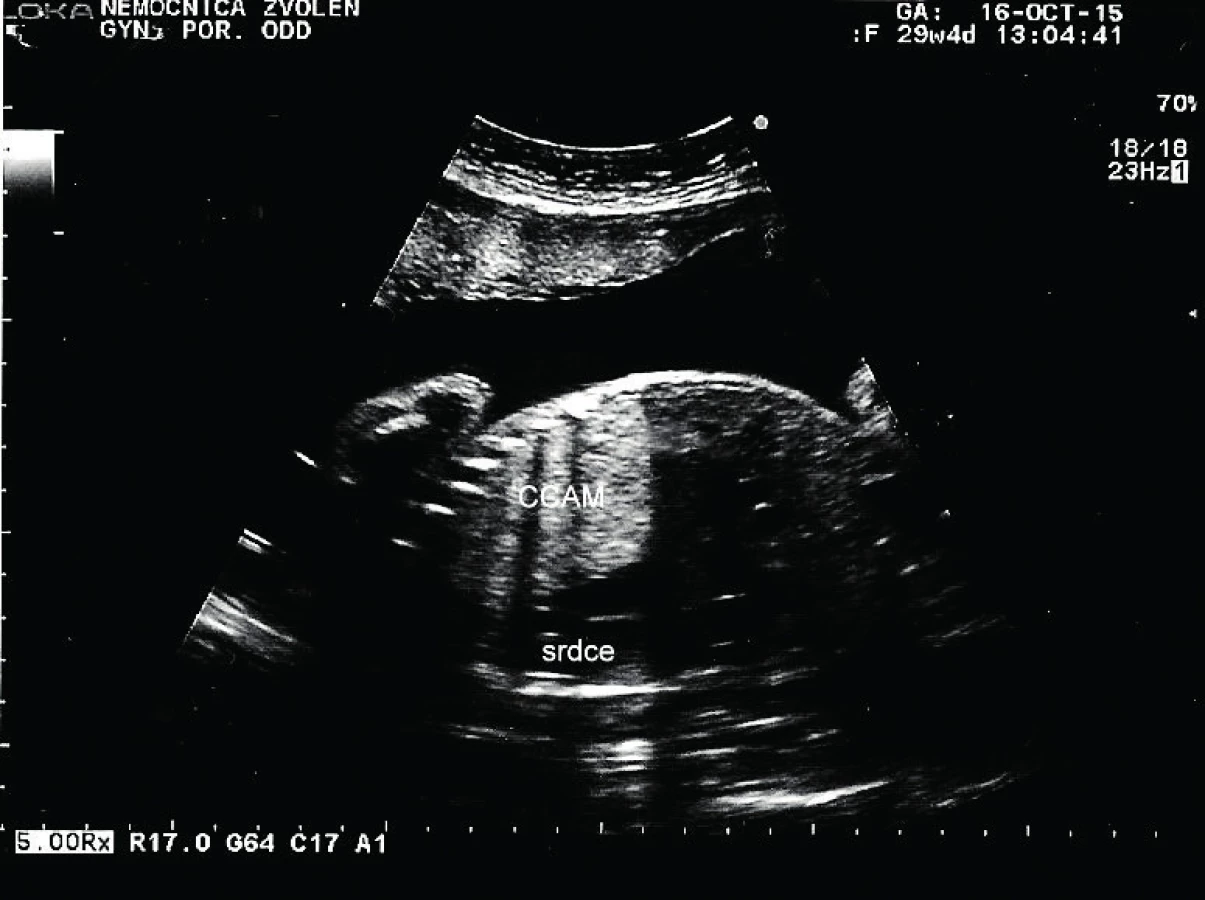
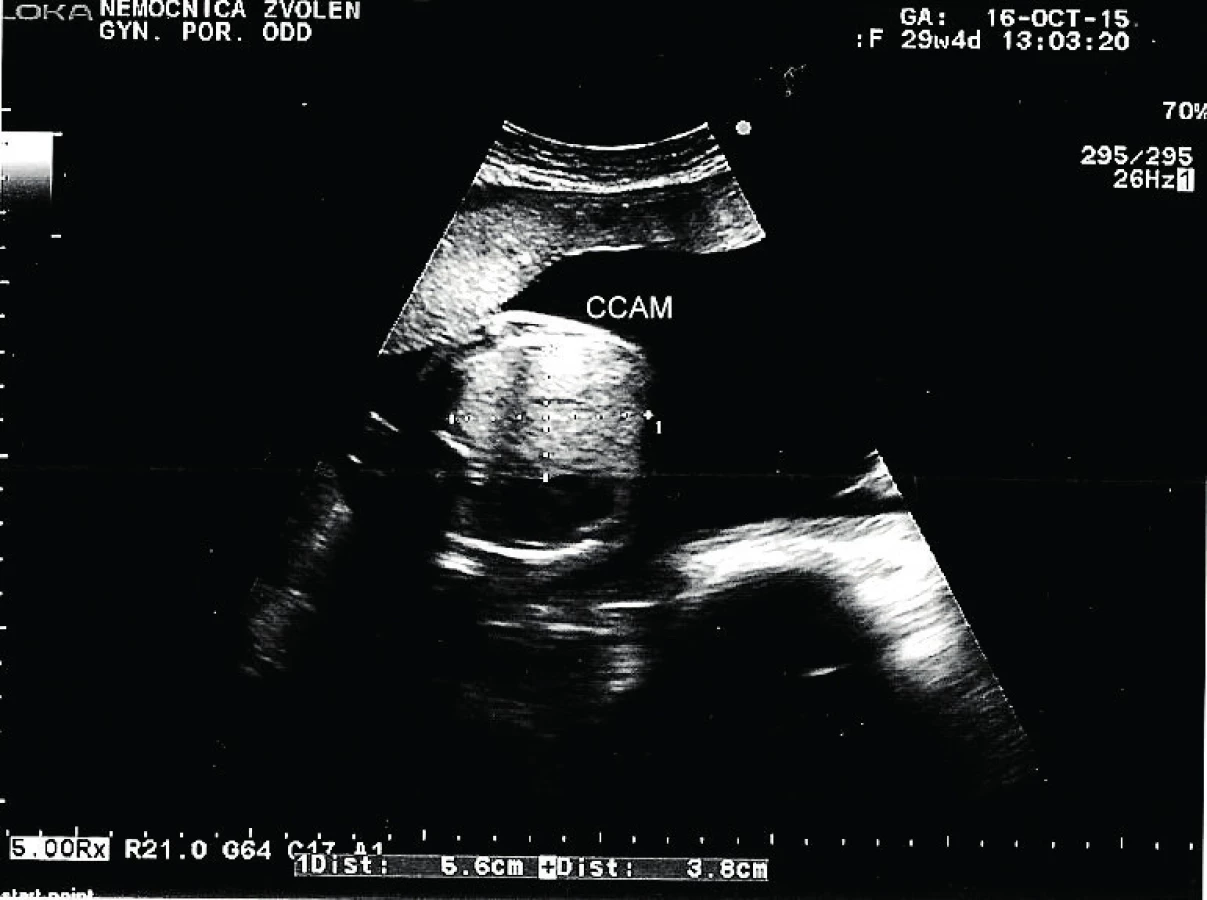
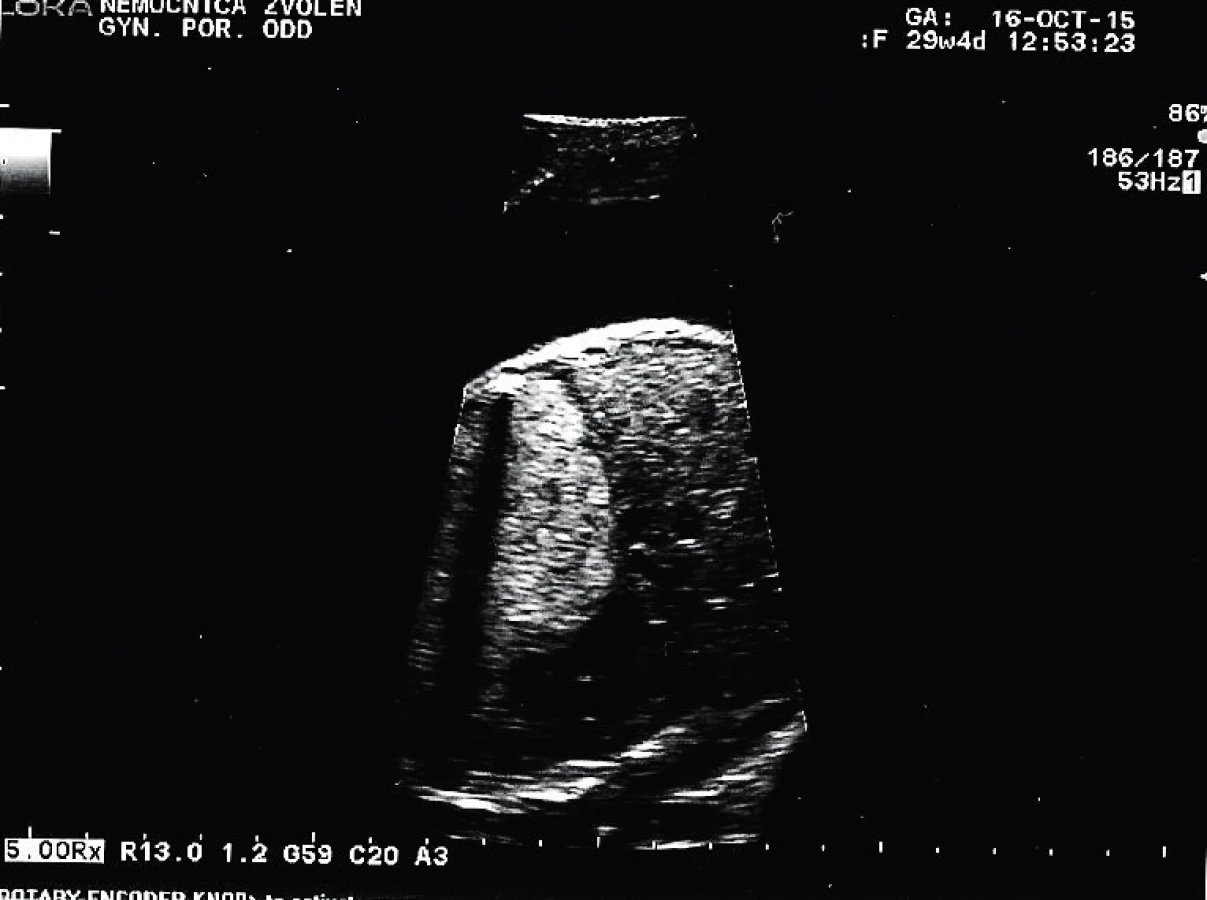
Tridsaťročná primigravida a nullipara sa dostavila na naše pracovisko v 31. týždni tehotenstva na prevedenie pravidelného ultrazvukového vyšetrenia v III. trimestri. Rodinná anamnéza bez pozoruhodností, v osobnej anamnéze bola prítomná autoimunitná tyreoiditída na terapii Euthyroxom 75 µg tbl p.o. Tehotenstvo prebiehalo bez komplikácií. Pri vyšetrení bol zistený polyhydramnion s AFI 29. Plod s hmotnosťou 1625 g, zodpovedajúci 30+2 týždňu. V pravom hemitoraxe plodu sa nachádzala výrazná homogénna hyperechogénna štruktúra veľkosti 59×41 mm. Ďalej bol viditeľný posun hrudných orgánov výrazne vľavo so zmenšením rozmeru srdca, C/T ratio 0,34, ale s normálnou anatómiou srdca. Bránica nad konkavitou pečene bola znížená, ale bez porušenia. Hydrops plodu nebol prítomný. Záver vyšetrenia: polyhydramnion, intratorakálna masa v. s. pľúcny sekvester s posunom srdca, t. č. bez známok kardiopulmonálnej dekompenzácie.
Pacientka bola odoslaná na spádové pracovisko vyššieho typu. Absolvovala superkonziliárne ultrazvukové vyšetrenie so záverom: diafragmatická hernia s posunom heparu do hrudníka. Bolo plánované vyšetrenie MRI a genetická konzultácia. V 31+2 týždni prichádza pacientka na príjem pre masívny predčasný odtok plodovej vody s pokročilým vaginálnym nálezom, bránka otvorená na 4–5 cm. Pre hypoxiu plodu, prejavujúcu sa na CTG zázname prítomnosťou intrapartálnych decelerácií II. typu, bol indikovaný pôrod akútnym cisárskym rezom. Dieťa sa narodilo o 19.07 hod. Apgar skóre bolo 3–3–7 a nasledovala intubácia. V hodine života sa dostavila cirkulárna instabilita a systémová hypotenzia. Aj napriek intenzívnej resuscitácii obehu a dýchania o 22.45 hod nastáva exitus letalis plodu. RTG vyšetrenie hrudníka odhalilo v pravom hemitoraxe cystickú prestavbu pľúcneho parenchýmu, ktorá sa správa expanzívne. Kontraktilita myokardu bola znížená. Patologické vyšetrenie placenty a pupočníka bolo so záverom ascendentnej infekcie – akútna chorionitída a chronická vaskulitída. Pitevný nález: kongenitálna cystická adenomatoidná malformácia pľúc vpravo, typ 3.
DISKUSIA
CCAM prvýkrát popísali autori Ch´in a Tang v roku 1949. Presná príčina vzniku tejto vrodenej vývojovej chyby (VVCH) nie je známa. Jedná sa pravdepodobne o inzult pôsobiaci v období medzi 7. až 20. gestačným týždňom a je prítomný nesúlad medzi rastom ciev, strómy a epitélií. Na bunkovej úrovni bol preukázaný nesúlad medzi proliferáciou a apoptózou [4, 21]. Rast lézie pokračuje do 25. týždňa a potom nasleduje plateau až regresia nálezov [7, 21]. Klasifikácia zahŕňa 5 typov, ktoré sa líšia na základe embryologického pôvodu a histologických vlastností [7, 21, 24, 25]. Typ 0 je najraritnejšia forma, ktorá vzniká z trachei a bronchov, postihnutie je ťažké a väčšinou letálne. Typ 1 je najčastejším typom (50–70 % prípadov). Prezentuje sa malým počtom veľkých cýst s rozmerom 3–10 cm. Je možný aj nález jednej veľkej cysty. Pôvod malformácie je z distálneho bronchu alebo proximálneho bronchiolu. Typ 2 pochádza z terminálnych bronchiolov a býva prítomný v 15 až 30 % prípadoch. V ultrazvukovom obraze sú viditeľné menšie cysty s priemerom 0,5 až 2 cm. U týchto plodov býva najčastejšie prítomná asociovaná anomália, ktorá má určujúci vplyv na prognózu plodu. Ide o srdcové vady (Fallotovu tetralógiu), agenézu až dysgenézu obličiek, atréziu gastrointestinálneho traktu a skeletálne anomálie. Typ 3 je prítomný v 5–10 % prípadov. Cysty sú mnohopočetné a malé, takže budia dojem solídnej masy a na ultrazvuku sa javia ako echogénna štruktúra. Pochádzajú z acínov. Typ 4 zahŕňa 5 až 15 % prípadov, opätovne sa jedná o cystické lézie s cystami väčšími ako 10 cm a býva asociovaný s malignitami, prevažne pleuropulmonálnym blastómom [7, 20].
Existuje ešte prenatálna ultrazvuková klasifikácia, kde sa podľa nálezu delia lézie na mikrocystické a makrocystické CCAM [3, 7, 8, 21]. Plody s mikrocystickými nálezmi majú v priemere horšiu prognózu [2]. V diferenciálnej diagnóze je nutné zvažovať bronchopulmonálnu sekvestráciu (BPS), diafragmatickú herniáciu, kongenitálny lobárny emfyzém, bronchogénnu cystu a teratóm. V prípade intratorakálnej BPS je rozdiel v krvnom zásobení, kedy CCAM je zásobený z pulmonárneho obehu a BPS má arteriálne napojenie priamo na aortu. Existujú aj výnimky, keď CCAM má napojenie na aortu, event. koexistuje zároveň s BP [5, 15]. Pri diafragmatickej herniácii je v hrudníku pozorovateľná peristaltika črevných kľučiek, vyprázdňovanie a napĺňanie žalúdka, event. presuny orgánov z hrudnej do brušnej dutiny. Mediastinálny teratóm v UZ obraze vykazuje viacnásobné tiene a v porovnaní s CCAM aj zvýšenú vaskularitu. Bronchogénna cysta pochádza z horných dýchacích ciest a jej odlíšenie od CCAM môže byť problematické. MRI je v sporných prípadoch uvádzané ako zobrazovacia metóda druhej voľby.
Manažment plodov s nálezom intratorakálneho tumoru zahŕňa referenciu na pracovisko vyššieho stupňa, kde by malo prebehnúť podrobné ultrasonografické vyšetrenie so zameraním na možné pridružené anomálie. Amniocentéza nebýva v diagnostickom postupe pravidlom [7].
Rodine je doporučená konzultácia s neonatológom a detským chirurgom. Pri náleze v nízkom gestačnom týždni je vhodné zváženie ukončenia gravidity, hlavne v prípadoch, keď má plod pridružené anomálie, abnormálny karyotyp, alebo javí známky zlyhávania cirkulácie. Pri menších nálezoch je možný pôrod v spádovej nemocnici. Väčšie a komplikovanejšie nálezy by mali byť centralizované, pretože je vyššia pravdepodobnosť nutnosti intenzívnej popôrodnej starostlivosti. Pôrod býva najčastejšie vedený per sectionem caesaream.
Medzi intrauterinné komplikácie patrí hydrops plodu, ktorý je spôsobený kompresiou vena cava. Znižuje sa venózny návrat a kardiálny výdaj [26]. Najčastejšie sa vyskytuje pri mikrocystickej forme CCAM. Veľmi raritne sa vyvinie po 28. týždni gravidity, pričom mortalita plodov býva vysoká. Ďalej sa z dôvodu obštrukcie ezofágu často pridružuje polyhydramnion. Z terapeutických možností máme k dispozícii torakocentézu, založenie torakoamniálneho shuntu, otvorenú fetomaternálnu operáciu s torakotómiou a lobektómiou, ex utero intrapartum treatment (EXIT) a predčasný pôrod. V prípadoch, keď tieto terapeutické možnosti nie sú k dispozícii, alebo nie je možné ich uskutočniť, existujú kazuistiky, kde sa po podaní steroidov prognóza plodu zlepšila [21, 28, 27]. Po stanovení diagnózy je doporučený monitoring plodov v týždňových až trojtýždňových intervaloch až do stabilizácie rastu tumoru, a potom sa frekvencia môže znížiť na jedenkrát mesačne. Ak plod začne vykazovať známky cirkulárnej instability až hydropsu, je nutné sledovanie zintenzívniť na niekoľkokrát týždenne [21].
Až 50 % plodov je postnatálne asymptomatických. V týchto prípadoch je doporučovaná plánovaná resekcia tumoru vo veku do 10–12 mesiacov života, ku ktorej sa pristupuje hlavne z dôvodu rizika malígneho zvratu, a taktiež sa zvyšuje šanca na ďalší vývoj pľúcneho tkaniva s následným zlepšením pľúcnej perfúzie, ktorá bola preukázaná pri kontrolách v 5 a 10 rokov po operácii [10, 19, 22]. V ťažších prípadoch sú prítomné príznaky respiračnej insuficiencie (od grantingu, tachypnoe až po fulminantné respiračné zlyhanie vyžadujúce ventilačnú podporu), kedy je nutné pristúpiť ku chirurgickému riešeniu hneď po pôrode ako život zachraňujúcemu výkonu [26]. Medzi postnatálne komplikácie patria dýchacie ťažkosti, opakované infekcie, hemoptýza, bolesti na hrudníku, dyspnoe, kašeľ a ďalšie nešpecifické príznaky. Príznaky sú následkom pulmonálnej hypoplázie, posunu mediastína, spontánneho pneumotoraxu a pleurálnych efúzií [26].
Prognóza závisí od typu lézie, histologickej štruktúry, pridružených anomáliách, prítomnosti hydropsu, alebo iných znakoch kardiopulmonálnej dekompenzácie a stupni pulmonálnej hypoplázie. Postnatálne prežitie je viac ako 95% [23, 28]. Plody, ktoré prekonali prenatálnu intervenciu, majú prežitie okolo 80 %. Pri plodoch s torakocentézou sa prežitie blížilo ku 100 % [28]. Jedna zo štúdií sa zaoberala dlhodobým neurologickým vývojom detí s mikrocystickým CCAM, ktoré prekonali fetálnu operáciu, a ich vývoj bol bez oneskorenia [1, 6].
ZÁVER
CCAM je málo častá vrodená vývojová chyba, ktorá môže mať rôzne klinické obrazy, oscilujúce od asymptomatického až po letálny priebeh, zahrňujúci aj intrauterinné úmrtia plodov. Pri prenatálnej diagnostike je nutné do diferenciálnej úvahy zahrnúť aj iné pľúcne anomálie a ich typické ultazvukové, respektíve MRI obrazy. Po prvotnej diagnóze je vhodné informovanie pacientky o prognóze a možnom vývoji anomálie a nutných popôrodných opatreniach. Ďalej je vhodné rozvrhnutie nasledujúcich ultrazvukových kontrol, aby sa monitoroval priebeh ochorenia. V prípade zhoršenia stavu plodu je vhodné zvážiť intrauterinnú intervenciu, v závislosti na dostupnosti terapie. V našom prípade bol diagnosticko-dispenzarizačný proces prerušený spontánnym odtokom plodovej vody a zhoršením stavu plodu, ktoré bolo tak závažné, že viedlo k nutnosti akútneho pôrodu cisárskym rezom s nasledujúcim úmrtím dieťaťa na podklade kardiopulmonálneho zlyhania.
MUDr. Kristína Bihariová
Gynekologicko pôrodnícke oddelenie
Nemocnica Zvolen a.s.
Kuzmányho nábrežie 28
960 01 Zvolen
Slovenská republika
e-mail: kika_s@seznam.cz
Sources
1. Adzick, NS., Harrison, MR., Crombleholme, TM., et al. Fetal lung lesions: management and outcome. Am J Obstet Gynecol, 1998, 179, p. 884–889.
2. Adzick, NS., Harrison, MR., Glick, PL., et al. Fetal cystic adenomatoid malformation: prenatal diagnosis and natural history. J Pediatr Surg, 1985, 20, p. 483–488.
3. Bunduki, V., Ruano, R., Marques DaSilva, M., et al. Prognostic factors associated with cystic adenomatoid malformation of the lung. Prenat. Diagn, 2000, 20, p. 459–464.
4. Cangiarella, J., Greco, MA., Askin, F., et al. Congenital cystic adenomatoid malformation of the lung: insights into the pathogenesis utilising quantitative analysis of vascular marker CD34 (QBEND-10) and cell proliferation marker MIB-1. Mod Pathol, 1995, 8, p. 913–918.
5. Cass, DL., Cromleholme, TM., Howell, LJ., et al. Cystic lung lesions with systemic arterial blood suply: a hybrid of congenital cystic adenomatoid malformation and bronchopulmonary sequestration. J Pediatr Surg, 1997, 32, p. 986–990.
6. Cass, DL., Olutoye, OO., Cassady, CI., et al. Prenatal diagnosis and outcome of fetal lung masses. J Pediatr Surg, 2011, 46, p. 292–298.
7. Sfakianaki, AK., Copel, JA. Congenital cystic lessions of the lung: congenital cystic adenomatoid malformation and bronchopulmonary sequestration. Rev Obstet Gynecol, 2012, 5(2), p. 85–93.
8. Gilbert-Barness, E. The respiratory system. In: Gilbert-Barness, E., ed. Potter´s pathology of the fetus and infant. St. Louis: Mosby-Year book, 1997, p. 712.
9. Gornall, AS., Budd, JL., Draper, ES., et al. Congenital cystic adenomatoid malformation: accuracy of prenatal diagnosis, prevalence and outcome in a general population. Prenat Diagn, 2003, 23, p. 997–1002.
10. Granata, C., Gambini, C., Balducci, T., et al. Bronchioloalveolar carcinoma arising in congenital cystic adenomatoid malformation in a child: a case report and review on malignancies originating in congenital cystic adenomatoid malformation. Pediatr Pulmonol, 1998, 25, p. 62–66.
11. Hubbard, AM., Adzick, NS., Crombleholme, TM., et al. Congenital chest lesions: diagnosis and characterization with prenatal MR imaging. Radiology, 1999, 212, p. 43–48.
12. Laberge, JM., Flageole, H., Pugash, D., et al. Outcome of the prenatally diagnosed congenital cystic adenomatoid lung malformation: a Canadian experience. Fetal Diagn Ther, 2001, 16, p. 178–186.
13. Laje, P., Liechty, KW. Postnatal management and outcome of prenatally diagnosed lung lesions. Prenat Diagn, 2008, 28, p. 612–618.
14. Lima, M., Gargano, T., Ruggeri, G., et al. Clinical spectrum and management of congenital pulmonary cystic lesions. Pediatr Med Chir, 2008, 30(2), p. 79–88.
15. Mandell, G. Imaging in congenital cystic adenomatoid malformation. http://emedicine.medscape.com/article/407407-overview#a1
16. Matsuoka, S., Takeuchi, K., Yamanaka, Y., et al. Comparison of magnetic resonance imaging and ultrasonography in the prenatal diagnosis of congenital thoracic abnormalities. Fetal Diagn Ther, 2003, 18, p. 447–453.
17. Mehta, AA., Viswanathan, N., Vasudevan, AK., et al. Congenital cystic adenomatoid malformation: a tertiary care hospital experience. J Clin Diagn Res, 2016, 10, p. SCO01-SC04.
18. Molinaro, F., Angotti, R., Garzi, A., et al. Prenatal diagnosis, 3D virtual rendering and lung sparing surgery by ligasure device in a baby with „CCAM and intralobar sequestration“. Open Med (Wars), 2016, 11(1), p. 200–203.
19. Murphy, JJ., Blair, GK., Fraser, GC., et al. Rhabdomyosarcoma arising within congenital pulmonary cysts: report of three cases. J Pediatr Surg, 1992, 27(10), p. 1364–1367.
20. Priest, JR., Williams, GM., Hill, DA., et al. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol, 2009, 44, p. 14–30.
21. Prima, FAFD., Bellia, A., Inclimona, G., et al. Antenatally diagnosed congenital cystic adenomatoid malformations (CCAM): research review. J Prenat Med, 2012, 6(2), p. 22–30.
22. Sauvat, F., Michel, JL., Benachi, A., et al. Management of asymptomatic neonatal cystic adenomatoid malformation. J Pediatr Surg, 2003, 38(4), p. 548–552.
23. Stanton, M., Njere, I., Ade-Ajayi, N., et al. Systematic review and meta-analysis of the postnatal management of congenital cystic lung lesions. J Pediatr Surg, 2009, 44, p. 1027–1033.
24. Stocker, JT. Cystic lung disease in infants and children. Fetal Pediatr Pathol, 2009, 28, p. 155–184.
25. Stocker, JT., Madewell, JE., Drake, RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol, 1977, 8, p. 155–171.
26. Stone, AE. Cystic adenomatoid malformation. http://emedicine.medscape.com/article/1001488-overview
27. Tsao, K., Hawgood, S., Vu, L., et al. Resolution of hydrops fetalis in congenital cystic adenomatoid malformation after prenatal steroid therapy. J Pediatr Surg, 2003, 38, 508–510.
28. Witlox, RS., Lopriore, E., Oepkes, D., et al. Neonatal outcome after prenatal interventions for congenital lung lesions. Early Hum Dev, 2011, 87, p. 611–618.
Labels
Paediatric gynaecology Gynaecology and obstetrics Reproduction medicineArticle was published in
Czech Gynaecology

2017 Issue 6
Most read in this issue
- Obrat plodu zevními hmaty z polohy podélné koncem pánevním po 36. týdnu gravidity – hodnocení úspěšnosti a komplikací
- Ambulantní miniinvazivní léčba funkčních cyst ovaria
- Kvalita DNA ve spermiích je negativně ovlivněna věkem mužů a je rizikovým faktorem početí
- Placenta percreta a její atypická lokalizace jako příčina silného nitrobřišního krvácení